Ultrafast infrared spectroscopy probes the character of the short, strong bonds in HF2–
In May 1919, an undergraduate chemistry student named Maurice Huggins at the University of California at Berkeley was panicking. His professor William Bray required his students to write a term paper to pass his course, but Huggins hadn’t yet done that, and the deadline was approaching. In desperation, he showed Bray rough notes he’d made about some of the ‘unsolved problems in chemistry’ that Bray had discussed. Might the professor accept these notes as the term paper, with a title added?
Bray did, but with caveats. ‘Huggins,’ he said, ‘there are several interesting ideas in this paper, but there is one you’ll never get chemists to believe: the idea that a hydrogen atom can be bonded to two other atoms at the same time.’1 Huggins had advanced this strange idea using the electron-sharing theory of chemical bonding proposed by Gilbert Lewis, depicting, for example, a dimer of hydrogen fluoride in which the four atoms were arranged in a square, with each hydrogen bonded to both fluorines. Lewis, however, was sceptical of the whole notion.
As is often the case, younger researchers were more ready to accept new ideas that older scientists resisted. So it was that in 1920 Wendell Latimer, a young lecturer at Berkeley, and the postdoc Worth Rodebush published a paper on Lewis theory in which they laid out Huggins’ suggestion, crediting him in a footnote.2 But this weird sort of bonding didn’t get a name until Lewis himself, having been won round, wrote in his seminal 1923 book Valence and the Structure of Atoms and Molecules that ‘the most important addition to my theory of valence lies in the suggestion of what has become known as the hydrogen bond’.
A century of debate
But the proposal by Latimer and Rodebush was largely dismissed or ignored for at least a decade; in 1926 British chemist Henry Armstrong ridiculed the idea that hydrogen could act as a ‘bigamist’, while in 1921 Irving Langmuir put forward a slightly different idea to explain how molecules such as water and hydrogen fluoride form dimers. Instrumental in the acceptance of the hydrogen bond was the intervention of Linus Pauling. In a 1925 paper with his graduate student Sterling Hendricks, he showed how such a bond might explain the ion HF2–, with the hydrogen sandwiched between fluorines. Pauling’s 1939 book The Nature of the Chemical Bond cemented the hydrogen bond as a part of the chemist’s lexicon, although he oscillated for a time between viewing it as a purely electrostatic phenomenon between ions or as a resonance between ionic and covalent forms. It wasn’t just Pauling’s status that secured the idea, but the fact that he tied it to crystallographic data.
Instrumental in the acceptance of the hydrogen bond was the intervention of Linus Pauling. In a 1925 paper with his graduate student Sterling Hendricks, he showed how such a bond might explain the ion HF2–, with the hydrogen sandwiched between fluorines. Pauling’s 1939 book The Nature of the Chemical Bond cemented the hydrogen bond as a part of the chemist’s lexicon, although he oscillated for a time between viewing it as a purely electrostatic phenomenon between ions or as a resonance between ionic and covalent forms. It wasn’t just Pauling’s status that secured the idea, but the fact that he tied it to crystallographic data.
Pauling’s uncertainty is understandable; although the hydrogen bond celebrated its centenary last year, the debate still hasn’t ceased. Students typically learn now that a hydrogen bond is essentially electrostatic: a positively polarised hydrogen attached to an electron-withdrawing entity such as oxygen or fluorine is attracted to an electron lone pair on another molecule. But it’s been long recognised that there’s some covalency in the hydrogen bond too.3 And while the hydrogen bonds in liquid water are about 25 times weaker than the covalent bonds in H2O molecules, others – like that in HF2– – are stronger and shorter, and generally regarded as delocalised, non-classical three-centre bonds.
Tipping point
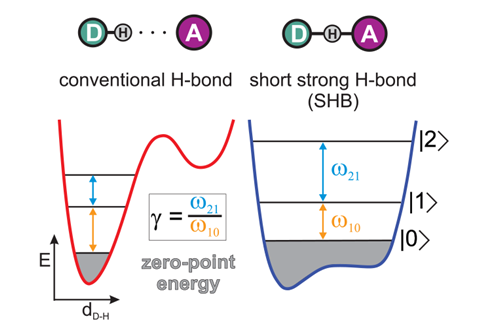
The latter bond is striking in that it can become symmetrical, with the hydrogen located at the midpoint of the fluorine atoms. While this was recognised decades ago, still this bond is being debated, as a recent paper by Andrei Tokmakoff of the University of Chicago and coworkers attests.4 They use ultrafast two-dimensional infrared spectroscopy to characterise the bonding in HF2– in aqueous solution, and see a smooth crossover from a conventional hydrogen bond (with the hydrogen located asymmetrically, covalently bonded to one fluorine) to a ‘short, strong’ hydrogen bond in which the hydrogen occupies a shallow double potential well. In the latter case the bond approaches a fully covalent character, but is strongly anharmonic in a way that mixes its bending and stretching motions.
In the extreme case – which both experiments and calculations show to have a significant lifetime – the hydrogen sits in a flat-bottomed single-well potential midway, on average, between the fluorines. Here the distinction between a hydrogen bond and a covalent bond vanishes: as the authors say, short, strong hydrogen bonds lie ‘at the tipping point where hydrogen bonding ends and chemical bonding begins’.
The same kind of structure can exist in principle in the so-called Zundel ion H5O2+ that hydrogen ions may form in water (as Huggins also acknowledged in 1971). But there it is extremely short-lived, and the spectroscopy is more complex. What’s more, quantum-mechanical tunnelling of the bridging proton in the double-potential-well structures becomes important even at room temperature, so that the Zundel ion can only be properly described with a full quantum description of the bonding rather than with classical notions of electrostatics or electron-sharing.5
In other words, here at least chemistry is shown to be a kind of coarse-graining of physics. It extracts useful heuristic concepts such as covalent, electrostatic and hydrogen bonding from the quantum molasses. But sometimes you just have to dip into the molasses itself.
References
1 D Quane, Bull. Hist. Chem., 1990, 7, 3
2 W M Latimer and W H Rodebush, J. Am. Chem. Soc., 1920, 42, 1419 (DOI: 10.1021/ja01452a015)
3 S J Grabowski, Chem. Rev., 2011, 111, 2597 (DOI: 10.1021/cr800346f)
4 B Dereka et al., Science, 2021, 371, 160 (DOI: 10.1126/science.abe1951)
5 F Mouhat et al., J. Chem. Theor. Comput., 2017, 13, 2400 (DOI: 10.1021/acs.jctc.7b00017)









1 Reader's comment