Clare Sansom takes a 'peak' at the databases that stop researchers being buried under an avalanche of chemical information
Clare Sansom takes a ’peak’ at the databases that stop researchers being buried under an avalanche of chemical information
With genome sequencing becoming routine, molecular biologists have a growing problem with data storage. Phrases like ’data mountain’ and ’data avalanche’ have peppered the bioinformatics literature for over a decade, and the issue is just as problematical now as at the turn of the millennium. Chemists have a data problem too, but ours is somewhat different. There are certainly a large and exponentially growing number of known compounds, but each entity (and therefore each database entry) is relatively small. As Antony Williams, founder and now vice president of strategic development for the RSC’s ChemSpider database, explains: ’Cheminformatics does not - yet - face the scalability issues of bioinformatics.’ Most chemical databases can be stored in their entirety on the hard drive of a fairly ordinary laptop or desktop computer.
But databases, particularly exponentially growing ones, are expensive to run. With a squeeze on public finances pulling in one direction, and an increasing expectation that scientific data should be free - or at least cheap to access - pulling in the other, it is not easy to come up with sustainable models for data access. The persistence of errors in chemical databases and their ease of use are also real issues.
At the start
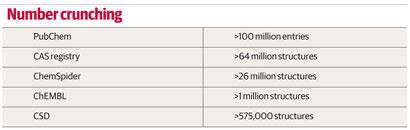
The era of chemical databases started in 1907 when the American Chemical Society launched, initially on paper and eventually online, its Chemical Abstracts Service (CAS) to catalogue the chemical literature. Its databases now hold entries for over 64 million compounds. The 60 millionth compound, a potential antiviral drug catalogued in 2011, came only two years after the 50 millionth milestone was hit - demonstrating the accelerating speed of novel molecule synthesis.
Three-dimensional structures of small (and medium-sized) molecules - ie chemical rather than biochemical structures - are held in the Cambridge Structural Database (CSD). Established in 1965 within the University of Cambridge, UK, the first edition of the CSD was again a book, containing references and tables of atom coordinates for a few hundred chemical structures.

The launch of the first software package for searching the CSD (the Quest software) in the late 1980s was quite a revolution, says Jason Cole, deputy director of the Cambridge Crystallographic Data Centre (CCDC), which maintains the database. ’It had something we would recognise today as a graphical user interface, and you could draw substructures and search with them.’ Today, the database contains over 575,000 structures, and still fits on a single hard drive. ’Our complete database takes up 100GB, which is small in bioinformatics terms, but what we send out to customers is of the order of 5GB,’ says Cole. The CSD uses a legacy data format, inherited from that needed by those early search algorithms. ’We are currently re-writing our database in a format fit for the 21st century. This will make our search engines faster, more powerful and easier to use, but it is the kind of unglamorous project that sits on the back burner for years,’ admits Cole.
Space to get in a muddle
If the records in all the different chemical databases were combined, removing duplicates - which might be an impossible task - the result would contain a couple of hundred million entries. It is quite instructive to ask what fraction of chemical space this represents. Several prominent chemists have predicted wildly varying numbers of possible chemical compounds; with Jean-Louis Reymond of the University of Bern, Switzerland, providing the most likely estimates. His group is attempting to answer the question ’how many small molecules are possible?’ by building a series of databases termed GDB [Generated Database (of Molecules)]. The latest variant, GDB-13, contains all chemically feasible structures with up to 13 atoms of C, N, O, S and Cl. ’Extrapolating Reymond’s numbers up to the largest possible drug-like molecules, with a molecular mass of about 500, would yield about 1018-1019 molecules,’ says John Overington, team leader for computational chemical biology at the European Bioinformatics Institute near Cambridge, UK. We have catalogued about one in 10 billion of these so far.

Most databases, apart from the CSD, only hold experimental data on the 2D structures and stereochemistry of molecules. This 2D data is often represented as simple text strings - called Smiles (simplified molecular input line entry specification) notation - to aid storage and searching. This type of representation is invaluable, but can also cause confusion. Not all research groups use the same interpretations and add-ons to Smiles, allowing different representations of the same compound to permeate the internet. Conversely, it can be difficult to show tautomers as essentially interchangeable forms of the same compound.
ChemSpider’s Williams tells an anecdote that indicates the limitations of the Smiles format. ’If you search for diamond in PubChem, one of the largest and most reputable chemical databases, you will retrieve the entry for methane,’ he says. ’This seems bizarre, but the explanation is simple: the Smiles representation of methane, CH4, is C - the same as for diamond or, indeed, graphite.’ The International Union of Pure and Applied Chemistry introduced the InChI (International chemical identifier) representation for compounds as a follow-on, rather than a replacement standard to Smiles in an attempt to reduce (rather than remove) these discrepancies.
Show me the money
The related issues of funding and data access is the elephant in the room in any discussion of scientific databases. A few decades ago almost every database was government funded. There are still many wholly publicly funded databases, particularly from US government and EU sources, but austerity measures and increases in the size and complexity of the databases are eating into this model from opposite sides. Cole from the CSD speaks for many in the community when he says ’grant agencies tend to think in terms of funding projects rather than funding resources, so it is much easier to get money to set up a database than to maintain it. Furthermore, with funding frequently offered for at most three years at a time, the database may often be put in jeopardy.’
Overington and his group at the European Bioinformatics Institute are building a free-to-access publically funded database of 2D structures and activities of pharmacologically active molecules. Their ChEMBL database is funded by the UK charity the Wellcome Trust, and contains data on around 1 million compounds, largely extracted from the literature. All our data have come from freely available sources, says Overington. ’As we focus on medicinal chemistry compounds with biological activities, most are fairly simple organic molecules,’ he explains.
The admittedly short history of one popular database, ChemSpider, demonstrates that it is possible to set up a database in a sustainable way without any initial funding. ChemSpider was started in 2007 by Williams - then chief scientific officer for Advanced Chemistry Development (ACD/Labs) in Toronto, Canada - initially as a hobby project with friends, with the vision of trawling the internet for chemical data and extracting it into a database. While the trawling never came to fruition, a first version of the ChemSpider database went live early in 2008 and was acquired by the RSC in May 2009. It now contains over 26 million entries, and the chemical community help with its expansion and curation using a crowd-sourcing model.

Using internet-based resources to build this database revealed a remarkable number of errors in reputable chemical sources, including five valent carbon atoms and clear mis-associations of chemical names and structures. This led Williams to a mission to improve data quality and to educate the community on which data sources are most reliable. He used a questionnaire on his ChemConnector blog to investigate which of eight widely-used public resources (including his own) was the most trusted, and tested the same resources for errors using the structures of 200 common small molecule drugs. He found errors in each database, including three serious ones in ChemSpider (immediately corrected using the web interface available to all users). The databases with the highest ’trust values’ were ChemSpider (perhaps unsurprisingly, since the survey was promoted via his blog) and PubChem. Interestingly, he confesses that he personally would never report that he always trusted a data resource.
That lack of absolute trust reflects an understanding that it will be impossible for any database to ever be 100% or even 99.99% reliable. Overington agrees that databases will always contain errors, but views this as rather less of a problem in practice. ’There is a hierarchy of representational errors and their subsequent impact in use - chemists typically view errors in formulas as the most important, followed by those in molecular graphs, stereochemistry, salt form and finally physical/crystal form. Different techniques are sensitive to different error types, and the impact of errors is context dependent - for example, for drug products, the crystal form can dominate product properties,’ he says.
Losing the cash cow
The CSD is one of relatively few chemical databases to have deliberately gone down a commercial route for funding. It was initially paid for by research grants, but by the late 1980s its management team became frustrated with the never-ending cycle of grant applications and concerned over the long-term sustainability of the resource. Believing - correctly - that the database contained enough information about pharmaceutically relevant compounds to interest the industry, they established the CCDC as a not-for-profit company. Over 80% of the income needed for the database is provided by industrial users and by the activities of a separate software company the CCDC operates.
But the pharmaceutical industry is changing. Until recently, many scientists have seen it as a cash cow, able to provide resources and cross-subsidise facilities for the poorer academic community. But with the cost per registered drug spiralling out of control, the boundaries between the two sectors are blurring. In a recent editorial in Drug Discovery Today, Barry Bunin and Sean Ekins of Collaborative Drug Discovery in Burlingame, US, discuss how collaboration with the public sector, and pooling data with academics and competitors, can help early stage drug discovery. Public domain databases such as PubChem and ChemSpider are cited there in a list of ’innovative tools [to] make drug discovery more efficient’.
The Collaborative Drug Discovery website allows users to store records of structure activity, pharmacokinetic and other data in a ’vault’. Bunin explains the company’s uniquely collaborative way of working: ’Users can choose to partition their data any way they want, maybe keeping some private and sharing other records within their organisation, with a group of collaborators or the entire community.’ Its business model as a for-profit company with a strong social focus on - and holding grants from the Gates Foundation and others for work on - neglected tropical diseases is also unusual. This example is further proof that the chemical community is still finding innovative ways to rise to the challenge of providing accessible and accurate data even in straitened times.
Clare Sansom is a science writer based in Cambridge, UK
Further Reading
B A Bunin and S Ekins, Drug Discovery Today, 2011, 16, 643 (DOI: 10.1016/j.drudis.2011.06.012)
Also of interest
![]() ChemConnector Blog
ChemConnector Blog
Helping to Create Connections in Chemistry






No comments yet