One route to developing new drugs is to look at targeting the hundreds of G-protein-coupled receptors that are not currently exploited clinically. Clare Sansom investigates
One route to developing new drugs is to look at targeting the hundreds of G-protein-coupled receptors that are not currently exploited clinically. Clare Sansom investigates
Nearly every medicine you take, be it a painkiller, an asthma inhaler or an antihistamine, targets the protein products of the human genome. Of course there are some exceptions to this rule, antibiotics and antivirals target the infecting parasite, and some experimental drugs are now targeting the machinery that creates those proteins.
Perhaps more surprisingly, 10 years into the human genome era the portion of the genome that is ’pharmaceutically interesting’ seems remarkably unchanged. For several decades, five groupings of proteins have been recognised as comprising the largest proportion of drug targets: hydrolases (including proteases), kinases, nuclear hormone receptors, ion channels and cell-surface receptors. Most important of all are undoubtedly the cell-surface receptors, which are the targets of around 60 per cent of drugs in current clinical use.
One group of receptor proteins in particular has risen to a position of pre-eminent importance to the pharmaceutical industry - the G-protein-coupled receptors (GPCRs). These are membrane-bound proteins that act as the cell’s communicators. Richard Henderson of the Medical Research Council (MRC) Laboratory of Molecular Biology in Cambridge, UK, an experienced researcher in membrane protein structure, describes them as ’signalling molecules that control the whole of physiology’.
Exact statistics are difficult to determine, but around half the profits of the pharmaceutical industry derive from drugs that target GPCRs. And the list of drugs that target these receptors includes some of the best-known and most widely used drugs of recent years, including triptans for migraine, beta blockers for hypertension, and Tagamet (cimetidine) for stomach ulcers. Other conditions that can be controlled by drugs targeting these receptors include psychiatric disorders, Parkinson’s disease and some forms of cancer. Yet until just a few years ago all the productive work in developing drugs to target this protein family had to be done without one of the most important weapons in the drug development armoury: detailed 3D structures of the proteins being targeted.
Almost 800 genes in the human genome code for GPCRs, although slightly over half of these are the pharmaceutically uninteresting olfactory receptors that enable us to smell. Fiona Marshall, chief scientific officer of specialist drug discovery company Heptares Therapeutics, based in Welwyn Garden City, UK, explains that the industry has done little more than scratch the surface of the others. ’Fifty six of the 370 non-olfactory GPCRs in the human genome are already targets of drugs in clinical use. However, several times that number are of interest to the pharma industry but have still not been "drugged". There are candidate drugs targeted to some further GPCRs in clinical trials, but many of these are large, lipophilic molecules that traditionally have a high failure rate,’ she says.
Scientists like Marshall and Henderson - who have devoted many decades between them to elucidating the structures and mechanisms of these proteins - believe that knowing what the GPCRs look like will help develop drugs to target the 300-odd currently undrugged GPCRs. In doing so, they hope to be able to fulfil previously unmet medical needs.
A clearer picture
There are two main reasons why GPCR structures have proved so difficult to determine. The first is that they are not simple to crystallise - they are membrane-bound, and cannot be purified and crystallised without further steps. Instead, membrane proteins need to be solubilised by detergents before being purified and persuaded - with difficulty - to form crystals.
While the first crystal structure of a soluble protein (myoglobin) was published in 1958, it took a further 27 years before researchers managed to crystallise and solve the structure of a membrane protein. The protein was the bacterial photosynthetic reaction centre and in 1988, Hartmut Michel, Johann Deisenhofer and Robert Huber were awarded the Nobel prize in chemistry for determining its structure. The recognition of their work came over a quarter of a century after the same prize was awarded to Max Perutz and John Kendrew for their investigations into the structures of the haem-containing globin proteins.
Stephen White of the University of California at Irvine, US, maintains a database containing the structures of membrane proteins and often compares its growth with that of the comprehensive protein structural database, the Protein Data Bank (PDB). ’The first version of our database was published in 1998, and contained fewer than a dozen proteins,’ he says. ’It now has rather over 600 structures, equivalent to the PDB in 1991, and these represent only 241 distinctly different proteins.’
The first low resolution structure of a transmembrane protein with seven helices was obtained in 1975. This was the prokaryotic light-sensitive proton pump, bacteriorhodopsin: an analogue of the GPCR family that captures light energy and uses it to move protons across the cell membrane out of the cell.
During the next 15 years, many developments in molecular biology and x-ray crystallography techniques laid the foundations that have made today’s detailed images possible.
The first ’true’ GPCR to yield to structural studies was the functional equivalent of bacteriorhodopsin: the light-sensitive protein rhodopsin. These proteins share a ligand, retinal, and a mechanism in which the cis-trans isomerisation of retinal induces a conformational change in the protein that couples to the G-proteins in rhodopsin and to proton pumping in bacteriorhodopsin.
A Russian group was the first to obtain crystals of rhodopsin. ’I remember a young PhD student showing me some pink needles when I was attending a FEBS [Federation of European Biochemical Societies] meeting in Moscow in 1983,’ says Henderson. ’These were rhodopsin crystals.’ In 2000, when Krzysztof Palczewski of the University of Washington in Seattle, US, Tetsuji Okada and Masashi Miyano from the Riken institute in Japan, and colleagues finally solved the structure of bovine rhodopsin to 2.8Å resolution, the floodgates were opened. This was not, immediately, for further experimental structures, but for structure prediction - ’homology’ or ’comparative’ modelling - using a more reliable model than the unrelated bacteriorhodopsin.
Unraveling structures
The rhodopsin structure showed that GPCRs consist of a bundle of seven membrane-bound α-helices, with a ligand binding site in the centre of the bundle and a binding site for a GTPase (the ’G-protein’) on the side that binds to the cell membrane. When a ligand binds to the receptor it induces the protein to change conformation into the active state, which leads to the exchange of guanosine diphosphate (GDP) for guanosine triphosphate (GTP) on the G-protein and the release of the GTP-bound G-protein from the GPCR. This then triggers a chemical response in an effector molecule inside the cell. GPCRs have evolved to bind to a wide variety of natural ligands and interact with many different effectors, and so can give rise to an enormous range of physiological responses.
The fact that each GPCR can exist in at least two conformations, ’active’ and ’inactive’ is another reason why determining the structure of GPCRs has, even among membrane proteins, proved particularly difficult. As White explains, ’GPCRs have to be able to flip easily from one conformational state to another -"off" to "on" - when a ligand binds. They are therefore less stable than most trans-membrane helical bundles, and this makes them almost impossible to crystallise with detergent in their native form.’
Leading the pack
During the last few years, the technical breakthroughs in this field have been pioneered primarily by three groups - Chris Tate’s at the MRC Laboratory of Molecular Biology and two based in California, US, led respectively by Ray Stevens of the Scripps Research Institute and Brian Kobilka of Stanford University.
The work of these teams has led to a step change in the prospects for GPCR structure determination. ’Since the first DNA and protein structures were solved in the 1950s, structural biology has progressed through a marriage between biology and technical development,’ says Stevens.
The techniques and methodologies developed by the groups are very different. Stevens’ and Kobilka’s work creates an extra hydrophilic domain by fusing a small protein into a long loop between two of the helices in the bundle. While Tate uses site-directed mutagenesis, altering each residue in turn, measuring the thermostability of each mutant and then combining the ’best’ mutations to make an optimally stable receptor locked in a pharmacologically defined conformation. All the groups have benefited from advances in microbeam x-ray diffraction that allow data collection from tiny crystals. ’A lot of membrane protein crystals are thin, flat plates that are difficult to work with,’ says White.

Kobilka and Stevens focused first on the structure of the β2 adrenergic receptor, the best characterised GPCR after rhodopsin and the target of the well-known asthma drug Ventolin (salbutamol). Kobilka, who identified the gene for this receptor in the late 1980s as a postdoc in Robert Lefkowitz’ lab at Duke University, US, provided the biochemical insights and expertise in protein engineering, with Stevens, who has also worked on GPCR structure determination for over 20 years, providing the necessary technological know-how.
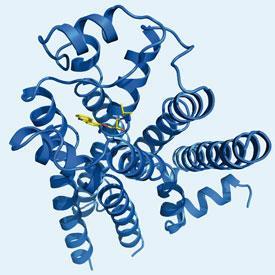
The first structures of the β2 adrenergic receptor GPCR were published in 2007 after the researchers had managed to ’lock’ the protein in an inactive conformation. This involved binding a small protein - either a lysozyme or an immunoglobulin domain - between the two helices, and a partial inverse agonist to the ligand binding site. Stevens’ group has also published structures of the human β2 adrenergic receptor bound to a number of different ligands and, in 2008, the A2A adenosine receptor. They have also determined structures for other medically important human receptors including the chemokine CXCR4 receptor, a proposed target for anti-HIV drugs, and the D3 sub-class of dopamine receptors.
Commercially viable
Two spin out companies have been fomed to use the structural know-how they have gained to develop new drugs that target GPCRs - San Diego, US-based Receptos was started by Stevens and Heptares was spun out from Tate’s group.
Receptos was principally set up to solve the human structures of, and design ligands for, a GPCR sub-family that binds the phospholipid sphingosine-1-phosphate (S1P). ’Targeting receptors in this family can meet previously unmet medical needs,’ says Stevens. ’Our primary target is the S1P1 receptor sub-type, an important target for multiple sclerosis, but we are also interested in targeting other S1P receptor subtypes for conditions including inflammatory bowel disease.’
Receptos’ first drug candidate, an S1P1 allosteric receptor agonist, is about to enter Phase I trials for multiple sclerosis and modulates the activity of the GPCR by binding to a site adjacent to the active site. ’Knowing the structures of different receptor subtypes allows us to design specific allosteric agonists and antagonists that bind to sites other than the natural ligand binding site. These sites differ between receptor sub-types more than the ligand binding site does, and targeting them can help reduce side-effects,’ adds Stevens.
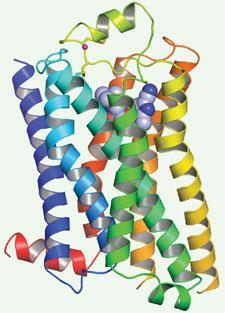
Tate, Gebhard Schertler and their colleagues solved the structure of the β-1 adrenergic receptor, an important drug target for hypertension, using site-directed mutagenesis to stabilise it in an inactive conformation. ’We were able to create a mutant adrenoceptor that was 400 times as thermostable as the wild type, by combining just six thermostabilising mutations,’ explains Tate.
In 2007, when they had obtained diffracting crystals, Tate’s team created Heptares to use the stabilisation technology they had developed to gain structural data on GPCR targets and design drugs that target them. Malcolm Weir, Heptares’s chief executive, explains his long association with the project. ’I was head of structural biology at GlaxoSmithKline in the early 1990s when that company first sponsored the Cambridge group to work on GPCR structures. At that time it seemed an intractable problem, which makes our recent progress seem even more remarkable.’
The company has been able to raise £21 million in venture capital funding even in the depths of recession. Its intellectual property is based on the patented ’stabilised receptor’ (Star) technology and they have now stabilised about 20 Stars covering 12 different receptors in active and inactive conformations. In fact, White, with his wide knowledge of the ’structural universe’ of membrane proteins, believes that scores of different GPCR structures are currently being worked on in industry.
Looking to the future
It may be useful to ask how these developments will affect GPCR based drug discovery in the long term: what will the industry be able to do with structures that it couldn’t before? Weir explains that atomic coordinates are not the only useful output from their new technological advances. ’Having reliable techniques for obtaining 3D structures of GPCRs is a major development, but this is not all we can do with our stabilised receptors. We can look at ligand binding directly using surface plasmon resonance and mutagenesis; measure binding kinetics directly; and use all this information to target drugs more precisely to allosteric binding sites. This should enable us to design more drugs to a wider variety of receptors, and make them more specific to receptor subtypes and so likely to have fewer side effects.’
Weir and Stevens believe that there is room enough in the field for both their companies and more. ’The GPCR family is extremely large and there is enough space in this structure arena for both our companies. We are competitors, yes, but we are collegial, respectful and friendly competitors,’ says Stevens.
Clare Sansom is a freelance science writer based in Cambridge, UK






No comments yet