

Small molecule manufacturer Toronto Research Chemicals helps drug formulators eliminate impurities in the development pipeline, ensuring safer products make it to market
Even in small quantities, pharmaceutical impurities can have serious negative health impacts and influence the behaviour and efficacy of a drug. This jeopardises the safety of patients. One research group found 71 out of 222 (32%) Food and Drug Administration (FDA)-approved novel therapeutics were impacted by safety concerns – designated post market safety events – between 2001 and 2010. There were three withdrawals, 61 boxed warnings and 59 safety communications, said the 2017 investigation.
Product recall due to the detection of impurities, or other safety reasons, can have major consequences for both consumers and pharmaceutical companies. Principally, patients are at risk of drug contamination, but producers must bear potential revenue losses and fines in mind. Public perception of the industry is a key factor too.
In 2018, the FDA reported that various angiotensin receptor blockers (ARBs) containing the active pharmaceutical ingredient (API) valsartan were found to contain N-nitrosodimethylamine (NDMA) and N-nitrosodiethylamine (NDEA) impurities. The report caused immediate alarm due to the potential hazards and sparked a product recall.
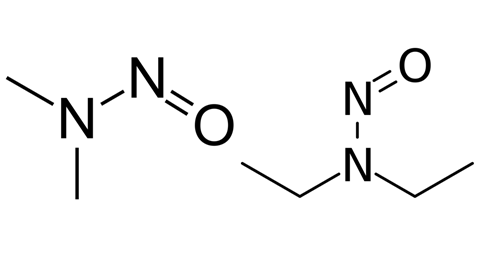
NDMA’s carcinogenic properties were first reported in 1956, NDEA’s in 1967. Like the wider nitrosamine family, they were classified ‘probable human carcinogens’ by the International Agency for Research on Cancer and the US Environmental Protection Agency. Despite there being traces in small quantities in food, the presence of nitrosamines in ARBs (used to treat hypertension, heart failure and kidney disease) caused widespread alarm.
Product recalls can have major consequences for both consumers and pharmaceutical companies
Subsequent recalls affected multiple therapeutics – including ARB batches containing valsartan, losartan and irbesartan – and has impacted other drugs in the years since. In April 2020, the FDA called for immediate withdrawal of all ranitidine (Zantac) products from the market, and voluntary recalls of diabetes medication metformin are currently ongoing.

The roots of the problem
A number of issues, including legislative failures, may have amplified the widespread pharmaceutical contamination by NDMA. Many of the therapeutics implicated have been generic, as are 90% of medications prescribed in the US. A market flooded with products from multiple manufacturers becomes harder to monitor, especially when that market is globalised. According to the FDA, just 28% of API manufacturing facilities for all US-sold drugs were located in the USA in 2019. Nearly one fifth were in India and 13% in China – double that of 2010.
2019 also marked a decline in the number of FDA inspections overseas with just 125 FDA inspections within China – 11% less than 2018 and 24% less than 2017 – a Bloomberg investigation showed. Since recalls impact products from large generic drug manufacturers globally, inspection regimes must be rigorous and ongoing testing by producers is crucial to protect consumers. Understanding how impurities form is vital to prevent them entering a final drug product. By accurately monitoring and preventing their formation, drugs with potentially harmful impurities will not reach the market. Impurities can arise at any stage of the drug formulation pipeline. Environmental factors – like temperature or humidity – can cause them as well as contamination from equipment, dosage form or drug degradation.
Organic impurity formation
Organic impurities fall into a number of categories. They can be degradants of the drug API or starting materials, unintended by-products of the manufacturing process or untransformed production intermediates. An API failing to separate from other materials involved in its synthesis – such as unreacted starting materials or intermediates – is one such instance. Impurities also occur when the by-products formed during synthesis do not completely separate from the active compound.
Instability in pharmaceuticals can also cause organic impurities to develop through API degradation over time. The FDA’s ranitidine recall for example, hinged on mounting evidence of a heat-unstable API.
Organic impurities are known to be harmful to human health, making strict limits on their presence in drug substances necessary
Many organic impurities, such as nitrosamines, are known to be harmful to human health and regulatory bodies and government set strict limits on their presence in drug substances. If thresholds are exceeded, qualification of the impurities can be necessary, resulting in costly toxicity studies. Robust quality assurance (QA) is the best defence against contamination by organic impurities. Regular testing of multiple product batches using reference standards – highly characterised materials that can be used to test the identity, strength, quality and purity of substances – should form part of QA process.
Inorganic and residual solvent impurities
Inorganic impurities, such as reagents, ligands, catalysts, inorganic salts and heavy metals used in vessels, filters and machinery, are easier to prevent and control. Using demineralised water and glass-lined vessels, and carefully monitoring each stage of the production process constitutes good practice.
Solvents used in the manufacture of a drug compound can be difficult to fully remove, meaning residues remain on the product. Solvents are therefore categorised according to their risk to human health, with strict limits set for safe daily exposure. Only the safest are approved for use in drug formulations and they require testing regimes to ensure continued consumer safety.
Depending on the complexity of the compounds – and the purity requirements in question – different combinations of techniques can be used

High-performance liquid chromatography (HPLC) with UV detection determines impurities for most pharmaceuticals. Quantitation can be performed versus an external standard of the impurity itself or by comparison to the response of the API. Depending on the complexity of the compounds – and the purity requirements in question – different combinations of techniques can be used.
Regardless of the pathway pursued, pure independently synthesised impurity samples are very useful. Matching the spectral and chromatographic profile of a detected impurity with those of a pure known reference provides a necessary benchmark to verify structure. Procuring a sufficient amount of an impurity allows it to be studied further, allowing full understanding of its toxicological effect and formation mechanism. This practice informs regulations that underpin impurity threshold safety.
At LGC, our TRC portfolio of pharmaceutical impurity standards supports drug development, medical and biomedical research professionals in more than 140 countries.
Our team of over 250 highly skilled chemists specialises in designing optimal synthetic routes for both new and known compounds, from impurities to customised chemical compounds and rare organics.
Case study: Synthesis of an atorvastatin impurity
The challenges surrounding pharmaceutical impurity analysis can be seen in the complex synthesis of the epoxy pyrrolooxazin tricylic (EPT) potassium salt impurity of atorvastatin.
This EPT salt impurity (A791895) is a photodegradation product of atorvastatin – a selective, competitive HMG-CoA reductase sold under the brand name Lipitor. It is the only drug in its class specifically indicated for lowering both elevated LDL-cholesterol and triglycerides in patients with hypercholesterolemia. The tricyclic impurity can cause adverse effects in users of the drug. As such, it is included in the mandatory list of impurities for which any drug manufacturer must test before they submit a new drug application.
Of all the impurities associated with the photodegradation of atorvastatin, EPT potassium salt is arguably one of the most difficult to prepare. To date, no synthesis has been reported in literature, making it difficult to obtain for use as a standard. We were contacted by a researcher who required EPT potassium salt, but was unable to find the molecule at a purity level appropriate for their analytical needs. We were tasked with developing a synthesis at the required purity which allowed isolation in quantity.
Despite its apparent complexity, the product was prepared in two steps from an advanced atorvastatin bicyclic impurity. However, the highly strained tricyclic molecule was susceptible to both heat and protic solvents (it slowly degraded even at 0°C), limiting our options both in terms of purification methods and reagents to effect the steps of the synthesis.
An alternate ether solution proved to be the optimal reagent to generate the desired K+ impurity from the methyl ester intermediate. Again, because of the limited stability of A791895 in a suitable solvent for NMR analysis, the structure of the impurity was verified indirectly via the ester precursor.
A 2D-NMR study comprising the dqfCOSY, NOSY, HSQC, HMBC-HC sequences (along with the usual H1, C13 and F19 NMR measurements) provided the essential information, allowing research labs to gain access to a supply of the EPT potassium salt at the appropriate purity level.
The impurity landscape is constantly changing. TRC is proud to support this vital research and safeguard the quality of therapeutics and wellbeing of consumers worldwide. What our teams do facilitates science for a safer world.











No comments yet