Researchers discover an overlooked chemical interaction in aspirin that could help understand protein structure
US based scientists have identified a previously unappreciated stereoelectronic effect in aspirin. This electron delocalisation sheds light on some of aspirin’s properties and could not only help in drug design but also be used to understand protein structure, say the researchers.
’We have been interested for several years in this n to pi* interaction,’ says Ronald Raines of the University of Wisconsin-Madison. That is, the delocalisation of an electron pair on a nucleophile to an antibonding pi orbital, often of a carbonyl.
Last year Raines published a paper claiming that these interactions could be found in almost all protein structures and could be as important as hydrogen bonding in the folding of proteins.1 ’But we were interested to see if these interactions went beyond protein structures,’ he adds.
So Raines went looking for a small molecule that had this interaction, so that they could characterise it in detail. He found it in the drug aspirin.2
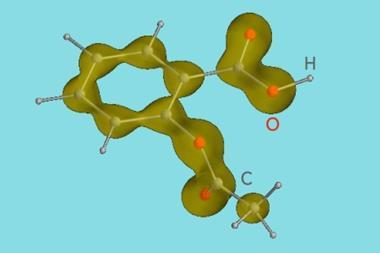
The interaction his group discovered occurs between aspirin’s carboxyl group and the neighbouring carbonyl moiety. ’Aspirin is perhaps the oldest of all drugs,’ he says. ’We examined its structure quite closely and we found that this n-pi* interaction seemed to be very strong in aspirin.’
This has some important implications. Firstly, Raines says that this interaction might be the reason that aspirin is such a good drug, because the interaction buries the carboxylate (the n-donor). This makes the molecule less polar, which Raines suggests could make the molecule’s passage to its target - through a ’hydrophobic channel’ - much easier.
But there’s also another reason that Raines likes aspirin, and it goes back to his work on the n-pi* interaction in proteins. Raines wants to develop some sort of test to detect the signature of this n-pi* interaction, which is much easier to do with a small molecule model system like aspirin, rather than using large biopolymers. Currently, this interaction is detected by observing a ’puckering’ in the crystal structure of the acceptor group, for example the carbonyl, ’but it’s hard to crystallise everything,’ he says.
’Simulations with classical force-fields do consistently result in insufficiently cooperative protein folding, relative to experiment,’ says Robert Best who works on protein folding at the University of Cambridge, UK. ’The interaction proposed by Raines could explain part of the missing energy, though is certainly not the only possibility.’
Properly understanding this electron donation, Raines says, will help understand why molecules take on the shape they do as well as how they interact with one another, just as the discovery of the hydrogen bonding helped to comprehend protein structure.
Laura Howes
References
1. G J Bartlett 6, 615( 2. A Choudhary, K J Kamer and Ronald T Raines, J. Org. Chem.






No comments yet