Chemists in the US have made important advances on two of the key challenges in the functionalisation of C–H bonds: how to selectively target a specific C–H, especially when it is geometrically inaccessible, and how to control and sequentially functionalise the notoriously difficult sp3 C–H bond. In achieving the latter, Jin-Quan Yu’s group at the Scripps Research Institute at La Jolla, California, have synthesised a library of synthetic chiral amino acids which could have applications in the pharmaceutical industry.
‘This new technique offers a very quick way to prepare unnatural amino acids, many of which are drug candidates or building blocks for peptide drugs,’ says Yu.
C–H bonds abound in organic chemistry, but they are tough bonds to break and control. Simple methods for functionalising these bonds could open up huge new vistas for organic synthesis. A typical strategy for cutting a C–H bond is to use a neighbouring functional group as a handle on which to attach a metal ion catalyst such as palladium. However, selectively attacking a C–H bond that is remote from the functional group is a problem.
Palladium crane
Yu’s team sought to direct palladium-mediated catalytic attack on the meta-C–H bonds of bicyclic anilines and benzylic amines. This was done by developing a series of bulky templates that are attached to the amine functional group. These act as the rigid boom of a crane, swinging the palladium to the further meta-C–H and avoiding the nearby ortho bond via a strained tricyclic cyclophane intermediate. Inserting a strategically placed fluorine atom into the structure can further enhance selectivity by acting as a ‘stopper’ to prevent the crane’s boom from taking an undesired ‘swing’. In this way, C–H groups up to 11 bonds away from the functional group can be activated.1
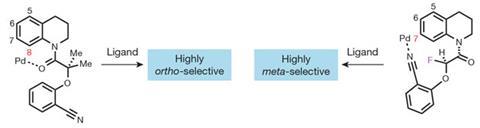
‘We believe this represents a versatile template approach to direct the remote meta-C–H bond activation of tetrahydroquinoline, benzoxazines, anilines, benzylamines, 2-phenylpyrrolidines and 2-phenylpiperidines, all of which are commonly used as building blocks in drug discovery,’ says Yu.
Daniel Morton, managing director of the NSF-funded Center for Selective C–H Functionalization in the US, says the new work is important. ‘With the ubiquity of C–H bonds found in organic molecules, selectivity is one of the intrinsic challenges to the field of C–H functionalisation,’ he says. ‘Directed C–H activation, using a functional group to guide the site of reaction, has focused almost exclusively on adjacent bonds, those five or six bonds away. With this report the Yu group take this science to the next level, with selective control over functionalization of C–H bonds as far as 11 bonds away. This has a huge impact on the scope of directed C–H activation, opening the door to novel substrates and new transformations.’
Unnatural amino acids
Meanwhile another team led by Yu has developed a way to control the arylation of a single (sp3)C–H,2 followed by functionalisation, with a different aryl group, of a second C–H bond on the same methyl carbon.
‘The challenge here is to cut the first C–H bond without the reaction proceeding to the second bond, and then cut the second bond to functionalise it with a different group,’ says Yu. The group discovered that a pyridine-based ligand promotes palladium-mediated monoarylation of primary ß-C–H bonds exclusively and that a second, quinoline-based ligand enables introduction of a distinct aryl group via subsequent secondary ß-C–H activation in one pot with high stereochemical selectivity.
Using alanine derivatives as a substrate, the researchers synthesised a range of unnatural chiral amino acids more quickly and simply than current methods allow. ‘Many carbon-hydrogen activation reactions that were once out of reach are now possible with these new ligands,’ says Yu. The laboratory is now working on second-generation ligands, seeking faster and more efficient reactions. ‘In general, we expect that these new developments will greatly expand the scope of research on unnatural amino acids as potential drugs,’ Yu says.
‘This study not only furnishes a method to produce chemicals of high value, but furthermore informs and leads the design of future catalyst systems, expanding the scope of transformations possible through C–H functionalisation,’ Morton comments.






No comments yet