Scientists in China have combined molecular dynamics with machine learning to carry out precise nanosecond-scale simulations of water ionisation,1 shedding light on a process that has been known for over a century but was tricky to understand because the events involved are so rare and hard to follow. ‘The extremely scarce probability of observing water ionisation under natural conditions obstructs direct experimental probing and poses a great challenge to the theoretical community for characterising the free-energy profile and revealing the reaction mechanism,’ says Ling Liu from Nanjing University, who is part of the team. He explains that although water autoionisation plays an important role in many chemical and biological processes, ambiguities surrounding the mechanism of this reaction have led to limited understanding of some of these events. The new findings could be used to study such systems in more detail, he adds.
‘The autoionisation of water has been a subject of extensive study since Arrhenius first proposed it in 1884,’ comments Gabriel Merino at Cinvestav-Mérida, Mexico, who wasn’t involved in the study. During the reaction, water molecules dissociate to form hydronium and hydroxide ions, but finding out exactly how that happens hasn’t been easy. ‘Accurate modelling of this process has posed a challenge,’ notes Merino. He believes that the results obtained by Liu and his colleagues are an important step in the right direction. ‘The essence of their achievement is accomplishing both time efficiency and high accuracy – made possible through well-tempered machine learning-assisted metadynamics – which allows the generation of nanosecond-scale free energy landscapes that result in the precise reproduction of two fundamental chemistry constants: the equilibrium constant and the autoionisation constant of water.’
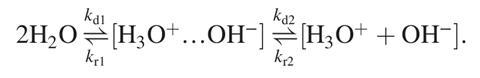
The team reported values of pKw=14.14 and kD=1.369 x 10-3s-1 for these two constants, respectively, both very close to experimental results. Liu points out that this is the first time that such close agreement between experimental data and modelling has been achieved and explains that this was possible because the new approach enables the characterisation of critical intermediate and transition states involved in the process. ‘Due to the limited number of molecular dynamics trajectories, previous studies lacked sufficient statistical sampling to mimic the complex ionisation pathway,’ he says, adding that the huge stability difference between water and the dissociated ions makes it impossible to obtain suitable data from conventional ab initio calculations.
‘To overcome this computational bottleneck, a machine learning technique was employed to replace the time-consuming DFT calculations,’ says Liu. He mentions that with the help of metadynamics, such large stability differences between reactants and products can be reduced by iteratively ‘filling’ the potential energy of the system, enabling frequent samplings throughout the whole reaction.
After analysing their results, the scientists concluded that the triple-proton transfer that initiates water autoionisation is a concerted process because they only observed one transition state located on the dissociation free-energy surface. ‘However, a strong asynchronous character was also confirmed, where the transfer of the third proton is obviously delayed,’ notes Liu. ‘This finding resolves disputes on whether water ionisation proceeds through double- or triple-proton transfer.’
Liu adds that water ionisation occurs through what he and his team call a dual presolvation mechanism, which starts with a pair of hypercoordinated and undercoordinated waters bridged by a water molecule. ‘This invokes an enhanced local electric field on the migrating protons and introduces a push–pull effect to promote water dissociation,’ he says.
Merino believes that the new results could have a significant impact in chemistry because the methodology ‘can be applied to a wide range of phenomena involving water and dissociation processes’.
References
L Liu et al, Phys. Rev. Lett., 2023, DOI: 10.1103/PhysRevLett.131.158001











No comments yet