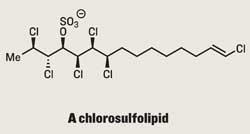
A chlorosulfolipid
A chlorosulfolipid
To the aspiring total-synthesiser, perhaps the most fruitful source of targets is the deep blue. Marine waters have produced some of the biggest celebrities of the natural product world - including the brevitoxins, saxitoxins and tetrodotoxins,1 famous as much for the sheer human effort needed for their landmark syntheses as for their complexity and size.
Far less documented, and until recently unsynthesised, are the chlorosulfolipids,2 also found wallowing in the sea, predominately in their phytoflagellate hosts. Like their more familiar marine neighbours, their key biological motif is their toxicity, resulting in the occasional case of seafood poisoning. However, this feature isn’t just of interest to gastronomes, as such molecules can be used to probe biological processes.
The name of these compounds gives away their major structural motifs - a generous helping of chlorine atoms arrayed on a lipidic bed, with a sprinkling of sulfates. Their similarity to polyketide natural products is remarkable, the hydroxyl groups simply switched for chlorine atoms. To the experienced organic chemist, this polychlorinated arrangement is somewhat terrifying, as many of the likely synthetic intermediates would be unstable - alkyl chlorides are prone to either elimination or even intramolecular cyclisation, so they must be designed with extra care.
Step by step
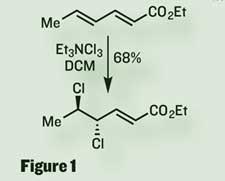
Erick Carreira of the Swiss Federal Insitute of Technology (ETH) was undaunted, though, and set out to make one of the more compact (and most prevalent - although still unnamed) members of this family.3 Well aware that the synthetic options were limited, his route uses an interesting array of reactions to build the target. The first key step, a dichlorination, sets the stereochemistry for the rest of the route (figure 1) - for which Carreira choose Charles Mioskowski’s tetraethyl ammonium trichloride.4 Treatment of a dienoate ester with this exotic reagent resulted in anti-dichlorination of the more electron-rich olefin in good yield - two chlorines down, four to go.
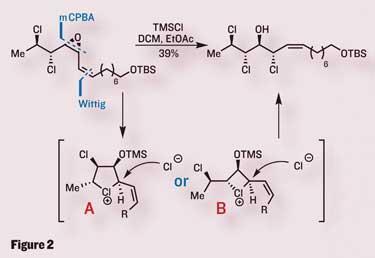
After elaborating this product, the team epoxidised the remaining olefin, ready to pop it open with trimethylsilyl chloride to install both the required chloride and hydroxyl. However, after a few more steps, and some hugely impressive NMR acronyms (if HETLOC and HECADE can’t help you, surely nothing will), it became clear that the stereochemical outcome of this reaction wasn’t the desired anti conformation expected of a simple nucleophilic substitution.
Carreira suggests that the syn product they observed instead could result if, before reacting with the attacking chloride, the epoxide had been opened by one of the two chlorine groups already present in the molecule (figure 2). If this cyclic chloronium intermediate was then opened by the incoming chloride ion, the syn product would form. Stereochemically, this is effectively a double inversion, or a net retention of stereochemistry - and required a rethink of strategy.
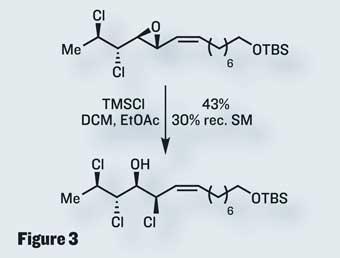
Hoping that this neighbouring-group participation would persist, the group produced an analogous starting material, this time containing a trans -configured epoxide. Using the same conditions, this epoxide opened with exactly the same ’double-inversion’, resulting in overall retention of stereochemistry, and the correct configuration for their synthesis (figure 3).
Use of Mioskowski’s reagent for a second time dichlorinated the remaining cis -olefin, with stereochemical control resulting from the substrate conformation. This syn -dichlorination gave the correct geometry in a cracking yield. Only one more chlorine atom was required, thankfully not at a stereocentre. This remaining sp2 centre was installed using a Takai-Utimoto olefination, completing the target in only ten steps. Not only is this a short and effective synthesis, it also opens up new strategies for the synthesis of the larger and more complex members of this intriguing group of natural products.
Paul Docherty is a medicinal chemist based in London, UK
References
Chem. Rev. 93 , 1897 Proc. Natl. Acad. Sci. USA 62 , 957 Nature 457 , 573 (DOI: 10.1038/nature07734) et al, Angew. Chem. Int. Ed. 36 , 2342












No comments yet