Aplykurodinone 1
Aplykurodinone 1
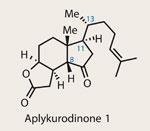
In the search for novel therapeutic agents, it seems that certain structural classes show up regardless of the target illness. Isolated in 1986, the steroid family of aplykurodinones have shown selective cytotoxicity in a variety of cancer cell lines, and add to the phenomenal list of steroids with potent medicinal properties.1,2
Organic chemists have been absorbed by steroids since their isolation and fascinating structure were first described. Many famous chemists have spent entire careers working on their intricately decorated polycyclic frameworks, but synthetic difficulties still persist. One notable challenge is controlling the ’freely rotating’ stereocentre on the side chain (numbered C-13 in the truncated aplykurodinone family).
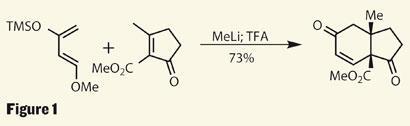
The team from Columbia University and the Sloan-Kettering Institute for Cancer Research, led by Samuel Danishefsky, has quite a history in the area of anticancer agents - as well as enviable synthetic prowess - so this was just the target for them to get their teeth into.3 A key step early in the synthesis is the ever-impressive Diels-Alder reaction using a rather complex (but possibly unsurprising) diene - the Danishefsky diene. This reactive, yet easily produced species is known for its synthetic utility, as a [4+2] addition into a cyclopentenone results in a cis -configured 6,5-ring system in great yield, and provides a useful enone functionality (figure 1).
A few steps later they added a very suspicious-looking diazoacetate - a group that’s desperate to lose a molecule of N2. Sure enough, treatment with an organocopper complex resulted in extrusion of nitrogen. The highly reactive carbene intermediate then added across the nearby double bond, constructing a complex trisubstituted cyclopropane (figure 2).
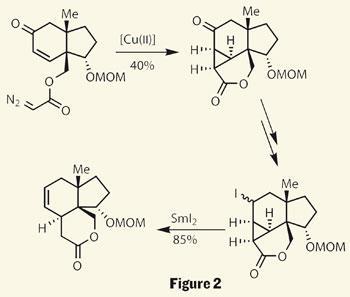
Examining the target, it’s perhaps a little odd that the group chose to install this three-membered ring, as it appears unnecessary. Their rationale becomes clear on treatment of the cyclopropane with samarium diiodide a few steps later, as the strained system cleaves open to reveal a 6,6,5-tricyle.
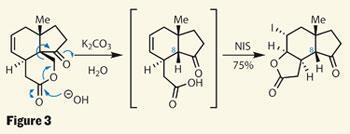
This still isn’t the target ring system, but it was all part of the group’s plan. They hoped to move the lactone ring around, extruding a one-carbon fragment en route. Danishefsky decribes this plan as ’traceless stereochemical guidance’, as the controlling element - the C-8 stereocentre and its methylene group neighbour - are removed in the process. This plan became reality in a hydrolysis and retro-aldol cascade, forming a carboxylic acid intermediate and releasing a molecule of formaldehyde. Better still, adding N -iodosuccinimide (NIS) triggered an iodolactonisation in the same pot, providing exactly the right ring skeleton (figure 3).
Finishing touches
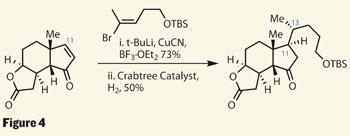
The group still had to take care of the side chain, and in particular, the C-13 problem. They managed to do the bulk of this in two steps - first was a Lewis-acid mediated conjugate addition of an organocuprate at C-11, to bolt on the side chain stereoselectively. The group followed this by reducing the remaining olefin to leave the C-13 position correctly configured. This is a common sticking point with hydrogenation of such steroids, where heterogeneous reactions tend to fail, so the group used the rather exotic (but homogeneous) iridium-based Crabtree catalyst rather than the more commonly used palladium on carbon (figure 4).
Three more steps were required to complete this target, finishing a rather complex target with deft control of stereochemistry. Further biological studies on this family of natural products may find some use for them in the clinic, but more importantly, the synthetic chemistry of steroids in general has been advanced.
Paul Docherty is a medicinal chemist based in London, UK
References
1 T Miyamoto, R Higuchi, T Komori, Tetrahedron Lett., 1986, 27, 1153
2 M J Ortega, E Zubia, J Salva, J. Nat. Prod., 1997, 60, 488
3 S J Danishefsky, Y Zhang, J. Am. Chem. Soc., 2010, DOI: 10.1021/ja1035495










No comments yet