Leiodolide B
Leiodolide B
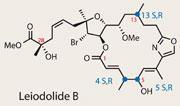
Although the story is incomplete, the target is a worthy challenge - leiodolides A and B have powerful activity and selectivity against NI60 tumour cells, and may lead to therapeutic agents. As is often the case, these molecules were found as vanishing traces in marine sponges - this time found skulking in the ocean at an appreciable depth of 220m. After drying and isolation, these compounds were received in minute quantities; 8mg of leiodolide A, and a paltry 0.8mg of leiodolide B. The astonishing reality of modern chemical analysis is that 0.8mg was sufficient for both biological and structural investigation - but unfortunately left some unanswered stereochemical questions.1
Stepping up to the crease was an extremely experienced team of macrolide-synthesisers lead by Alois F?rstner of the Max Planck Institute for Coal Research in M?lheim, Germany. They were looking to both crack the stereochemical quandary, and try out some neat organic synthesis en route. Key challenges include a tetrahydrofuran moiety containing five adjacent stereogenic centres, one of which is quaternary.2
Ringing the changes
Their synthesis began with that complex tetrahydrofuran ring, building it from simple precursors (figure 1). After a simple enyne synthesis, treatment of the allylic alcohol with titanium isopropoxide, hydrogen peroxide and an asymmetric salen ligand resulted in enantioselective epoxidation of the alkene. Similar to the more frequently used Sharpless asymmetric epoxidation, this protocol developed by Tsutomu Katsuki from Kyushu University in Japan has a particular affinity for cis -substituted olefins, as found in this example - and delivers fantastic yield and stereoselectivity.3
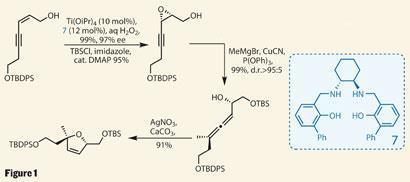
Adding a cuprate - formed in situ from methylmagnesium bromide - performed a conjugate addition into the alkyne rather than attacking the epoxide directly. This formed an allene, and was again achieved with impressive control of stereochemistry. Finally, treatment of the allene with silver nitrate and calcium carbonate closed down the desired five-membered ring in the form of a dihydrofuran, leaving both stereocentres intact. Further functionalisation of the remaining alkene using N-bromosuccinimide in aqueous dimethylformamide appended the last two stereocentres around the ring.
Stereochemical frustration
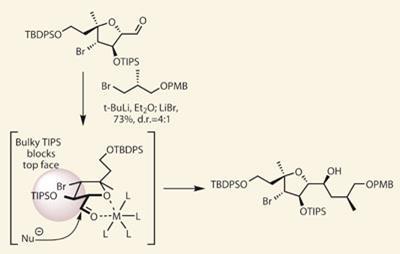
The next challenge was to find a way to couple this fragment to the rest of the molecule, and to set the problematic C13 stereocentre in both possible configurations. The team decided that the best way to be entirely sure about that stereocentre was to define it within a pre-prepared fragment, diverging their synthesis at this stage into two routes - one where the product would be 13S, and the other 13R. Unfortunately, the bulky triisopropylsilyl (TIPS) protecting group got in the way when they attempted to alkylate the relevant aldehydes. However, using a metal to tie the aldehyde into a more accommodating position allowed the nucleophile to be steered past the steric block (figure 2).
I lack the space on this page to discuss the rest of the synthesis in detail, save to say that the group created three further fragments. When correctly assembled, these completed several variants of the suggested structure for the methyl ester of leiodolide B, varying the stereochemistry at three key positions (C13, C4 and C5). Expecting that some combination of these configurations would match the target, the team were clearly devastated to find small but significant differences scattered throughout the 13C-NMR spectra.
Although they failed to find a perfect match, the team remains confident that the consitution - that is, the gross connectivity of the atoms - of leiodolide B is correct. Their plan is to synthesise the more abundant leiodolide A, whose structure is more confidently assigned, and attempt a biomimetic interconversion. A grand plan, but one which we’ll have to wait some time to appraise.
Paul Docherty is a science writer and blogger based in Reading, UK
References
1 J S Sandler et al, J. Org. Chem. 2006, 71, 8684
2 A Larivée et al, Angew. Chem. Int. Ed., 2010, DOI: 10.1002/anie.201005850
3 Y Sawada et al, Angew. Chem. Int. Ed., 2006, 45, 3478










No comments yet