Nakadomarin A
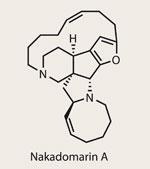
Every year, hundreds of good total syntheses are published with a fantastic key step, generally showcasing methodology developed by the same group. What turns a good synthesis into a great synthesis are the steps surrounding that motif, something that Darren Dixon from the University of Oxford, UK, exemplifies with this synthesis of Nakadomarin A.
This target is a relative youngster, isolated just over a decade ago from a marine sponge (a curiously plentiful source of grant-material for synthetic chemists and biochemists alike). Whilst not particularly potent as a potential pharmaceutical, it’s breadth of activity is surprising, encompassing anticancer cytotoxicity, kinase inhibition and both antibacterial and antimicrobial aptitudes. Plenty of excuses to tackle that hexacyclic 8,5,5,5,15,6 alkaloid ring system, then; indeed, enough for this to be the fourth synthesis this decade.
Have a little patience
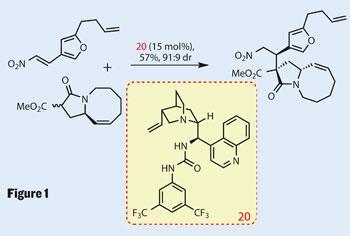
The chemistry really gets interesting after the synthesis of the two main building blocks, which each take around five steps to make. One is a 1,3-dicarbonyl compound, similar to a malonate, in which the acidic central position is racemic. The other has an unsaturated nitro moiety perfectly set to accept a nucleophile. Their Michael-type coupling, using a fairly complex (cinchona-derived) organocatalyst (Figure 1), is particularly impressive, creating two new stereocentres, one of which is quaternary - resulting from setting of that acidic centre. The only fly in the ointment is the reaction time of eight days - but good things come to those who wait...
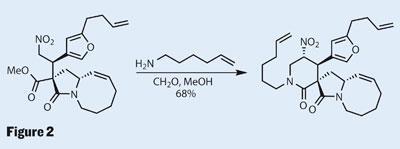
More carbon atoms were required to complete the target, and these were introduced directly, in a ring formation developed by Dixon. His method is to combine two reactions in a cascade: firstly, a nitro-Mannich reaction, followed closely by a lactamisation (Figure 2). As its name suggests, the nitro-Mannich is simply a Mannich reaction (addition to the iminium ion formed by formaldehyde and an amine) in which a nitro group forms the nucleophile - acting much like a carbonyl group to provide sufficient electron-pull to form an enolate-like anion.
Having completed its task, the nitro group was now redundant, and was removed using ’a modification of the Ono procedure’. I hadn’t read this procedure before - using a little azobisisobutyronitrile (AIBN) and tributyl tin hydride, the group were able to cleanly remove the nitro group under typical radical conditions. The simplicity of this method, and its usefulness in enolate-type chemistry mean that it’s likely to turn up in my lab book rather soon.
Packing in the complexity
The final two steps in the synthesis accomplish a considerable amount; having successfully reduced the larger lactam in preference over the smaller γ-lactam, it was the turn of the smaller. A little diisobutylaluminium hydride (DIBAL-H) partially reduced the lactam to an iminium ion, which reacted with the nearby furan to form the final five-membered ring stereoselectively (Figure 3). This completed the core of the molecule, with only the macrocyclic 15-membered ring left to install.
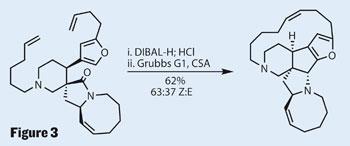
Such large rings often show only a small cis/trans bias, normally in favour of the trans isomer. However, Nakadomarin A requires a cis relationship, which has been a stumbling block in previous syntheses. Interestingly, Dixon found that a small amount of acid could overturn the trans selectivity in the ring-closing metathesis. This, the group presumes, protonates the tertiary amine group, and thus prevents any interaction of the nitrogen lone pair with the Grubbs generation 1 catalyst. Unfortunately, the diastereomeric ratio could not be pushed above 2:1, requiring a high performance liquid chromatography separation to divide the two alkene isomers.
Completing the synthesis in only 12 steps (following the longest linear sequence) is quite an achievement for such a complex little target, and represents one of the most elegant syntheses I’ve read in 2009.
Paul Docherty is a medicinal chemist based in London, UK
References
, 4267 (DOI: 10.1021/ol801666w)
4 J Tormo, D S Hays and G C Fu, J. Org. Chem. 1998, 63, 5296












No comments yet