Platencin
Platencin
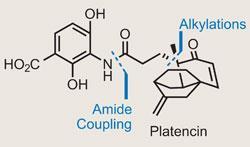
The need to discover new antibiotics to treat resistant strains of bacteria is a well- documented and discussed challenge for chemists. This is an area of medicinal chemistry that over the past 100 years has leant heavily on natural products, including neomycin, vancomycin and the cephalosporins. Development of new treatments waned in the last few decades of the 20th century, but the recent emergence of multi-drug-resistant bacteria has sparked renewed efforts, a good example of which is platencin (and close family member, platensimycin).
Both structures were unearthed by an isolation team at US pharmaceutical firm Merck, and were quickly found to have an inhibitory effect on bacterial fatty acid biosynthesis (FAS), with a particular penchant for drug-resistant strains.1 As if that wasn’t enough, the proposed structures of these targets are fascinating to the synthetically-minded - the right
hand side (RHS) fragment of both features a complex polycyclic cage that immediately lays down the synthetic challenge (figure 1). Syntheses of platensimycin have come in thick and fast - the first racemic work by K C Nicolaou at the Scripps Research Institute in La Jolla, US, was followed by a further nine formal syntheses.2
However, the more therapeutically useful platencin has had only two total syntheses, and two formal. Recently, two rapid, enantioselective approaches to platencin have joined this cadre, both sharing the same disconnection to a key RHS fragment required for the formal synthesis, and both featuring a cycloaddition reaction at their core.
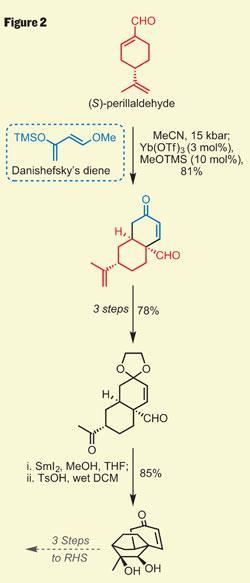
The first of the two - winning the race to publish by a single day - was completed by Floris Rutjes of the Radboud University, Nijmegen, in the Netherlands.3 His approach was to start from a commercially available terpene, perillaldehyde (figure 2). This chiral building block already contains one stereocentre, which was used to direct a Diels-Alder addition of ’Danishefsky’s diene’, a well-known and exceptionally useful substrate.4
However, reactions of such cyclohexenes with this diene are known to be sluggish, and neither heating nor catalysis could persuade the two to react efficiently. But Rutjes remembers his undergraduate thermodynamics quite well, and put the reaction under pressure, quite literally. This installed a second ring, leaving the team with one further cyclisation to complete.
A few steps were required to elaborate the substrate, including a ’protection’ of the aldehyde by Wittig olefination, and then ’deprotection’ via a selective ozonolysis, leaving the required bis-carbonyl functionality for a pinacol coupling. Using the ever-versatile samarium diiodide (which I’m sad to report I’ve never used - the shame!), the final six-membered ring was formed in excellent yield. Three more steps completed the RHS fragment, and the formal synthesis.
Pipped to the post
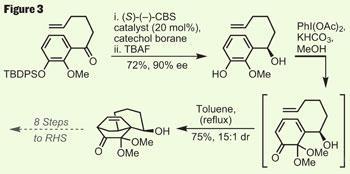
Just a day after Rutjes, Nicolaou - who has an almost unnatural obsession with these targets - published his eighth route into this family in three years, and again a Diels-Alder cyclisation is the critical step (figure 3).5 Shunning nature’s chiral building blocks - to an extent, as all asymmetry involves nature somewhere - his route involves a CBS (Corey-Bakshi-Shibata) reduction of a pro-chiral ketone. This gave the key substrate for the Diels-Alder in excellent enantiomeric excess, generating the controlling stereocentre for the cyclisation. The diene required for the cycloaddition was generated in situ by a hypervalent iodine oxidation - an exchange of solvents and some thermal encouragement triggered the desired cyclisation in good yield and selectivity. However, a further nine steps were required to elaborate this structure to the same RHS fragment.
Both routes have their advantages, but the real benefit is that both encourage development of such targets into drug candidates. And perhaps that leaves us - the bacteria-susceptible - as the real beneficiaries.
Paul Docherty is a medicinal chemist based in London, UK
References
Nature 441 , 358 et al 45 , 7086 Angew. Chem. Int. Ed. 47 , 6576 et al 96 , 7807 J. Am. Chem. Soc. , 2008 (DOI: 10.1021/ja804588r)












No comments yet