Dirk Trauner's team at UC Berkeley has completed an elegant biomimetic synthesis of Rubioncolin B, using a key tautomerisation and intramolecular Diels-Alder sequence that mirrors the proposed biosynthetic pathway and offers insights into natural product formation.
Impersonating nature isn’t easy, and biomimetic syntheses are remarkable in two senses. First, the chemist has to make the (often tricky) intermediate, or analogue thereof, thought to be present in the biological process. Then, reaction conditions thought to be similar to those in nature have to be recreated - not always easy, as nature seems to shun even the shiniest quick-fit round-bottomed flasks. However, the biosynthetic insights these reactions reveal, and their often astounding synthetic grace, mean this type of total synthesis will always be popular.
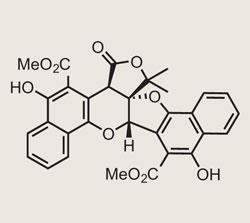
Dirk Trauner of the University of California, Berkeley, has quite a history with the Rubiaceae family of natural products, and their biomimetic synthesis.1 Their use in traditional medicine2 makes them appealing from a biological activity view-point, but I’d rather think it’s their remarkable structure that holds his attention. His retrosynthetic analysis of Rubioncolin B3 hinged on a key tautomerisation / intramolecular Diels-Alder (IMDA) sequence from an analogue of a proposed biosynthetic intermediate. Both reactions in this sequence are reversible - but molecular modelling of each intermediate suggested their desired target was the most stable, so the equilibrium should lie convincingly on the right side.4
Quick split
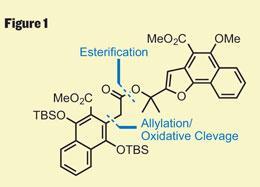
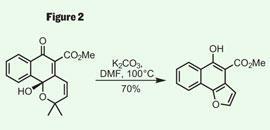
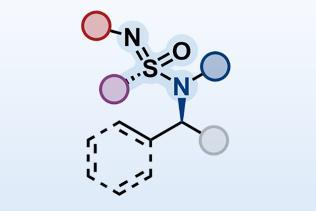
With these ambitious steps planned, the more immediate problem was the synthesis of the precursor (figure 1). This is often where the organic chemist’s impersonation of nature comes unstuck, as these substrates can be as synthetically challenging as the target itself. However, Trauner’s approach was concise - splitting the molecule into halves of comparable complexity, he was able to quickly assemble both and stitch them together with an esterification. His synthesis of the ’right-hand’ fragment uses a particularly neat ring-contraction - starting with a dihydropyran, opening with base and then re-closure of the ring resulted in the furan required after expulsion of a molecule of acetone (figure 2).
Ringing the changes
With the synthesis of the protected precursor complete, it was time to challenge their biosynthetic proposal. Trauner’s team chose an unusual reagent combination to deprotect and oxidise the substrate. In place of the more commonly used TBAF (tetrabutylammonium fluoride) or hydrofluoric acid, they used TASF (tris[dimethylamino]sulfonium difluorotrimethyl silicate), a non-basic source of fluoride. This was essential as the more common reagents decomposed the substrate due to their strongly basic or acidic properties. Also interesting was the use of a hypervalent iodine oxidant (iodosobenzene diacetate in this case), which is mild, selective, and compatibile with TASF.
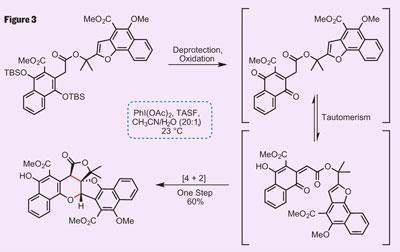
Combining these reagents gave the required reactive substrate, which reversibly tautomerised to form an ortho -quinone, as expected (figure 3). This tautomer then completed the IMDA reaction, forming two new rings and setting three stereocentres into exactly the configuration found in the natural product. This intermediate only required a little elaboration to complete their synthesis - cleavage of the methyl ether - finishing a remarkably elegant route to the target. Moreover, the success of their biomimetic approach lends considerable insight into the biosynthetic origin of this class of natural products.
Paul Docherty is a medicinal chemist based in London, UK
References
1 D Trauner et al,Org. Lett., 2005, 7, 5865
2 R Singh et al,Chem. Biodiversity, 2004, 1, 1241
3 H Itokawa et al,Chem. Pharm. Bull., 1993, 41, 1869
4 D Trauner et al,J. Am. Chem. Soc., 2008, DOI: 10.1021/ja803498r







No comments yet