Vannusal B
Vannusal B
To those readers who’ve a little experience with total synthesis and its literature, a glance ahead to the first structure will already have seen the proverbial cat leaving the bag. The key is in the compound’s title: ’Originally Postulated Structure’ . This is a classic case of misassigned identity - the structure published by the researchers who first isolated the compound from its natural source has been recreated via total synthesis, and found wanting.
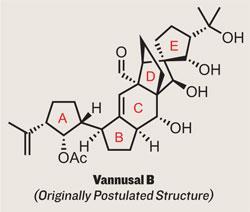
In this case, the intrepid researchers tackling the synthesis were under the direction of Professor K C Nicolaou, working in his labs at the Scripps Research Institute near San Diego.1 A man of quite some experience in this field, he’s certainly come across natural products with an identity crisis before, but I imagine that the discovery after
many years of synthetic effort must still feel shocking.2
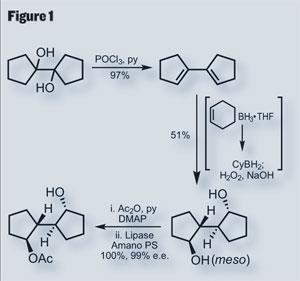
Unaware of these looming complications, Nicolaou began the retrosynthesis by splitting the molecule into two (almost) equally sized halves, to be united about the ’C’-ring. The lower portion, containing the ’A’ and ’B’ rings, had a rather impressive genesis starting with an achiral diol featuring several axes of symmetry (figure 1). A bis -chlorination and elimination then gave a diene, which was oxidatively hydroborated. To form the correct regioisomers from this hydroboration, they used a slightly bulky reagent, cyclohexyl borane. The resulting diol has four stereocenters, but is of meso configuration as there is still symmetry embedded in the molecule. This symmetry was finally broken by acetylating both alcohols, and then treating the bis -acetate with a lipase enzyme, which selectively chewed off one acetate to give the optically active target in both excellent yield and enantiomeric excess.
The second half
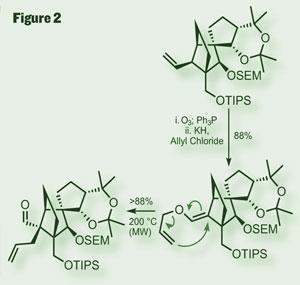
Focusing on the other portion of the molecule (part of which was published back in 2002), the paper describes work progressing from a reasonably advanced intermediate.3,4 While one was already in place, a second quaternary stereocenter was required, for which they turned to a Claisen rearrangement. This venerable reaction is approaching its centenary, but is still often on A-team in total synthesis (that’s as much of a sporting analogy as I’ll ever achieve), thanks to its disregard of steric congestion, and the frequently predictable stereochemical outcome. In this case, an allyl-vinyl ether, constructed with ease by ozonolysis and O -alylation of its enolate, was heated in the microwave to promote a thermal [3,3] sigmatropic rearrangement. Not a new idea by any measure, but inspiring (figure 2).
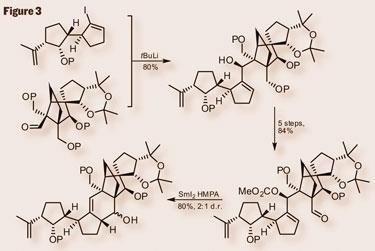
The coupling of the two fragments used a Shapiro reaction to install a vinyl iodide, lithium-halogen exchange and then addition to the aldehyde partner giving the coupled product (figure 3). However, the result was nice, with only a single diastereoisomer produced. The team then needed a few steps to exchange protecting groups, oxidise a primary alcohol and form a secondary carbonate. This intermediate was then ready to cyclise, forming the ’C’ ring by using samarium diiodide to generate a radical anion from the aldehyde. Allylic attack by the radical, and expulsion of the carbonate completed the carbocyclic skeleton.
A mixture of determination and skill allowed the group to take both isomers forward to the target. However, disaster befell them when comparing the synthetic material to the spectral data reported for the naturally isolated sample, as there wasn’t a match.5 Nicolaou states that the spectra were similar, but unfortunately no suggestion as to an alternative structure is yet forthcoming. However, if any researcher has the resources and experience to tackle this thorny problem, it’s Nicolaou.
Paul Docherty is a medicinal chemist based in London, UK
References
et al, Chem. Commun., 2002, 2480
4 K C Nicolaou et al, Angew. Chem. Int. Ed., 2005, 44, 3874
5 G Guella et al, Angew. Chem. Int. Ed., 1999, 38, 1134












No comments yet