Paul Docherty comes face to face with an old adversary
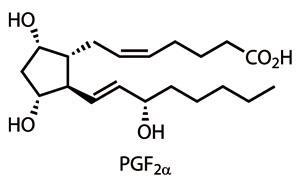
The rationale for all this effort is mostly due to their function as mediators in every animal system, including humans.1 Their roles are diverse – from vasodilation to bronchoconstriction – presenting a phenomenal target for medicinal chemists. Indeed, several synthetic prostaglandins are marketed, including treatments for pulmonary hypertension, peptic ulcers and to induce childbirth.
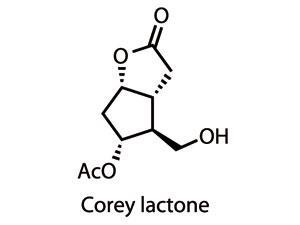
Recognising that the bicyclic heart of the Corey lactone is fundamental in controlling the stereochemistry of later steps, a small team led by Varinder Aggarwal at the University of Bristol, UK, has now devised an inventive and phenomenally short route to a variant of this well recognised bicycle.3 Starting from 2,5-dimethoxytetrahydrofuran, hydrolysis in water provided succinaldehyde, a reactive dialdehyde.
Treating this with (S)-proline resulted in initial dimerisation to an adduct, now bearing three aldehyde moieties and a hydroxyl (figure 1). This species remains in equilibrium with a lactol, featuring only two aldehyde groups, which can participate in a dehydrating aldol reaction to generate the desired target.
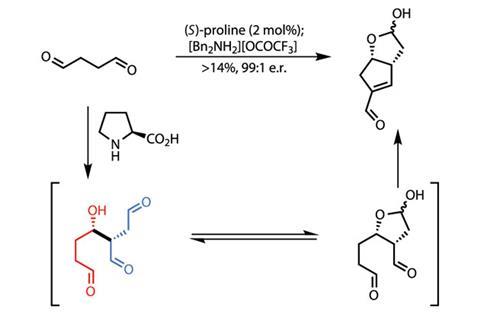
However, all those aldehyde groups present in all four species allow many competing processes to occur, which limits the isolated yield to only 14%. This may appear restrictive, but in setting two stereocentres in high yield and diastereoselectivity, as well as creating two rings, the team has achieved an enormous, cheap and scaleable shortcut over previous methodology.
Simple methylation of the lactol protected this fragile functional group. This freed the team to focus on the single remaining aldehyde, which they treated with a complex organocuprate reagent (figure?2).4 This chemistry delivered the first of two sidechains required for any prostaglandin synthesis with excellent control of diastereoselectivity. By trapping the product as a silyl enol ether, the team could then use reductive ozonolysis to remove the extraneous carbon. This installed the required hydroxyl group on the cyclopentane ring, and with the required stereochemistry by virtue of substrate control.
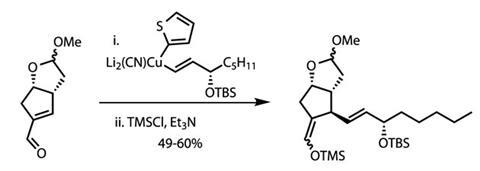
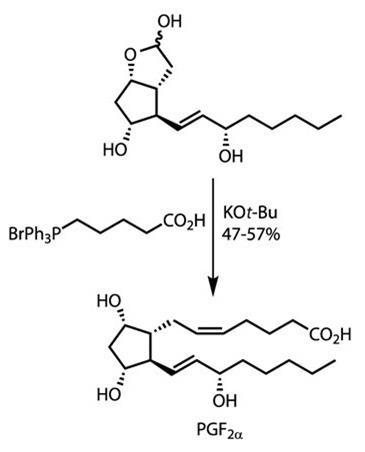
This left the team with only one further sidechain to install, and they did this with ease. Partial hydrolysis of the acetal removed the methyl protecting group, freeing the lactol functionality. When exposed to a Wittig reagent bearing the required sidechain, a smooth reaction returned gram quantities of the completed target in an astonishing seven steps (figure 3).
Not only does this synthesis constitute a phenomenal route to an important natural product, it paves the way to smarter, shorter and cheaper syntheses of potentially medically useful analogues of the prostaglandins. The team in Bristol should be very proud of their efforts – but I have to admit a little envy!
Paul Docherty is a science writer and blogger based in Reading, UK
References
1 K C Nicolaou, E J Sorensen, Classics in total synthesis, 1996, VCH, p65, ISBN 978-3527292318
2 E J Corey et al, J. Am. Chem. Soc., 1969, 91, 5675 (DOI: 10.1021/ja01048a062)
3 G Coulthard, W Erb and V K Aggarwal, Nature, 2012, 489, 278 (DOI: 10.1038/nature11411)
4 B H Lipshutz et al, J. Organomet. Chem., 1985, 285, 437 (DOI: 10.1016/0022-328X(85)87388-5)












No comments yet