After years languishing behind more fashionable areas of research, the main group elements are back in vogue. James Mitchell Crow finds out why
It is fair to say that main group elements are not the most glamorous members of the periodic table. The s-block metals such as calcium, and p-block elements like boron and germanium, are literally outshone by their flashier transition metal neighbours gold, palladium and rhodium. Even in pure chemistry terms, the reactivity of main group elements has always seemed a little staid compared to the virtuoso catalytic tricks that many transition metals routinely perform. As early as the 1970s, it seemed that the main group elements had already given up all their secrets, leaving future generations of researchers with little new to find.
‘Within inorganic chemistry, main group was the most boring part of my undergraduate degree,’ recalls Cameron Jones, a researcher at Monash University in Melbourne, Australia, who completed his degree at the University of Western Australia in 1984. ‘At that time, it was thought that everything we needed to know about the chemistry of the main group elements was known: the properties, oxidation states and reactivity were pretty well developed and there were no new areas to go,’ he says. Transition metal chemistry, in comparison, was undergoing a period of explosive growth that is still to let up.
But at the same time as Jones was learning the dry conventions of classical main group chemistry, the first signs of a renaissance in the field had already begun to appear in the literature. In the early 1980s it was discovered that p-block elements such as silicon and phosphorus could be forced into exotic states of existence – low oxidation and low coordination states – within compounds that were perfectly stable at room temperature. Although potential applications would have been far from the minds of the chemists first discovering this wild new frontier of main group compounds, more recent work suggests that their chemistry has a richness – and a usefulness – to rival that of the transition metals that have long overshadowed them. And Jones is among those researchers currently at the forefront of the resurgence.
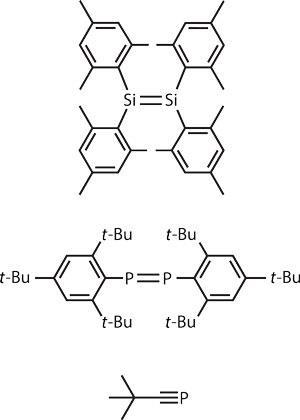
1981 is regarded as the turning point, Jones says. In that year, three disruptive discoveries were published that began to break apart the main group chemistry orthodoxy – starting with the so-called double bond rule, which stated that only the lightest members of the periodic table would form stable double bonds. Robert West at the University of Wisconsin–Madison in the US, and his colleagues were the first rule-breakers, creating the earliest example of a stable compound incorporating a silicon–silicon double bond, the equivalent of an alkene in organic chemistry.1
Soon after, Masaaki Yoshifuji at the University of Tokyo in Japan and his team made the first phosphorus–phosphorus double bond.2 And in the same year, Gerd Becker and his co-workers at the University of Stuttgart, Germany, synthesised a compound incorporating a phosphorous–carbon triple bond.3 In the latter compound, the phosphorus was not just in a low oxidation state, it was also one-coordinate, hinting at a future in which main group elements with vacant coordination sites might act as catalytic centres able to capture substrates for catalysis.
Bulking up
The key to this breakthrough was ligand design. These three compounds are united in their use of bulky substituents to stabilise the main group element in question in a state that would otherwise be too unstable. ‘Steric bulk allows kinetic stabilisation of low coordinate and low oxidation state systems,’ explains Jones. ‘It hinders them from reacting with oxygen or water, or undergoing disproportionation processes [which would return the element to its more usual oxidation states]’. West’s disilene, for example, featured a total of four bulky, benzene-based ligands called mesityl groups, whereas Yoshifuji used two even bulkier tri-tert-butylphenyl groups to create his diphosphene.
As bulky ligand design allowed more and more stable examples of main group elements with low oxidation state or coordination number, increasing numbers of researchers began to join the field. ‘It probably wasn’t until the turn of the millennium that it really took off – now it seems that nearly every Nature Chemistry or Science issue has got something on it,’ Jones says.
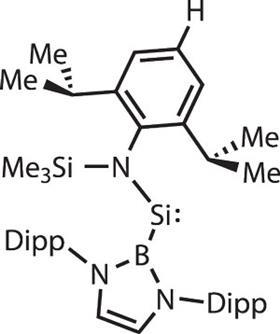
The examples of stable compounds featuring main group elements in states once thought impossible continue to roll in. Not so long ago, stable acyclic silylenes – the silicon equivalent of a carbene, a highly reactive species that is only two-coordinate and which features an electron lone pair on the silicon – were unheard of. In 2012, Jones and his collaborators at the University of Oxford and University College London in the UK published the first example, stabilised by a bulky boron-based ligand.4 ‘It actually ties in nicely with West’s first disilylene,’ he says. ‘The way he made it was to start with a [conventional] silicon(iv) compound and photolyse it. He suggested that he made a silylene intermediate, which was a deep blue colour, stable only below 77K, and then that dimerised to give the silicon–silicon double bond.’
Like Jones, Matthias Driess was still a student when the pioneering work of West and Yoshifuji was published. ‘Their breakthrough, demonstrating that you could isolate these compounds, put them in a bottle and study their reactivity – at this stage I was fascinated,’ he recalls. Driess now works as a researcher in this area at the Technical University of Berlin in Germany – an area undergoing a shift from simply making these new compounds to trying to put them to use. ‘After people had hunted new species, they asked themselves “What can we do with them?”’ he says. ‘After a certain time playing around with new systems, once you recognise what they can do, you would like to use them for the benefit of mankind. For main group chemistry, this has just started.’
The development that pointed to a way that low oxidation state main group compounds could be put to use was the realisation around the middle of the last decade that they are capable of activating small molecules – such as hydrocarbons, dihydrogen and carbon dioxide. ‘The dream is to use non-metals as catalysts for the important transformations of small molecules that we all rely on,’ Driess says.
Move over, metal
Why go to the trouble of developing main group-based catalysts? Transition metals are the well-established masters of catalysis chemistry, and new facets to their reactivity continue to be unearthed. But they also have a drawback, points out Doug Stephan, a researcher in main group catalysis at the University of Toronto in Canada who has perhaps pushed main group catalysis the closest toward synthetic utility (see box). The most active transition metal catalysts tend to be the expensive precious metals, such as rhodium, palladium and platinum. These metals are also considered toxic, which causes a problem when they are used to synthesise pharmaceuticals, for example. ‘One of the biggest costs associated with the production of drugs these days is the removal of transition metals,’ Stephan says. Switching to main group catalysts would ease this problem, he says – and boron or silicon, say, are also significantly cheaper than any precious metal. Other transition metals such as iron or nickel also hold this appeal, he admits – the main group systems simply offer another alternative.
Driess’s dream of metal-free catalysis is one shared with many others in the field, Jones included. ‘People are looking more and more at the reactivity, and these are very reactive systems – that, I think, is the next stage of the renaissance of main group chemistry,’ he says. Jones’s recent publication revealing the first acyclic silylene, for example, also showed that the compound would activate dihydrogen even below room temperature – the first silicon compound shown to do so.
Using a main group compound to activate dihydrogen – breaking the hydrogen–hydrogen bond in an oxidative addition process, so that the two hydrogen atoms both end up covalently bonded to the silicon atom – was first achieved by Phil Power and his team at the University of California, Davis in the US. Power used a digermyne, the germanium equivalent of an alkyne.5 The process may sound trivial, but is in fact no small achievement. ‘Dihydrogen is the smallest molecule you can think of but with the strongest covalent bond between two homoelements,’ Driess says.
Whether main group or transition metal, an element needs several key features to be able to pull dihydrogen apart in this way. ‘The centre needs a vacant coordination site – an empty orbital into which to suck up the electrons of the dihydrogen,’ says Driess. To do that effectively, the energy of this orbital (the lowest unoccupied molecular orbital, or LUMO) must fairly closely match that of the dihydrogen donor orbital (its highest occupied molecular orbital, or HOMO). ‘That’s what transition metals beautifully fulfil – a vacant coordination site, and a LUMO that matches the energy of the HOMO of the dihydrogen molecule,’ says Driess. ‘But that is also the beauty of some main group low coordinate compounds,’ he adds. ‘Today we have a lot of examples of silicon compounds with a vacant coordination site – now we have to learn how to tune the electron level, the LUMO acceptor level. We are just at the stage where we can show that this is really possible.’
As with West and Yoshifuji’s work, the key to accessing these new compounds is all in the ligand design. Steric bulk is a prerequisite – but too bulky and the incoming dihydrogen molecule will not be able to access the central silicon atom. Similarly, the ligand’s chemical make-up influences orbital energy levels. ‘At the moment, some of the best ligands we have are nitrogen-chelating ligands called beta diketaminatyl ligands,’ says Driess. ‘And then we have carbenes. I think these are the star ligands at the moment; the N -heterocyclic carbene ligand is really one of the breakthroughs in main group chemistry.’
As an example, Driess points to the work of Guy Bertrand at the University of California San Diego in the US, who has been making boron compounds in which the central boron atom is flanked on each side by a N-heterocyclic carbene (NHC).6 Boron is typically a quintessential Lewis acid: an atom blessed with only three electrons in the neutral state, it is a long way short of the eight needed for maximum stability, and so is known as an excellent electron acceptor. However, NHCs are such powerful electron donors that the boron atom itself switches behaviour to become electron-donating. ‘It’s turning the world upside down,’ says Stephan, whose eye has also been caught by the work. ‘It’s taking a Lewis acid and turning it into a Lewis base. It’s fundamental research at this stage, but terrifically exciting,’ he adds.
Learning to cycle
Although developing low-valent main group compounds that can successfully coordinate small molecules such as dihydrogen has now been achieved, this step is just the first in a sequence of reactions that must be developed if Driess’s dream of turning main group compounds into useful catalysts is to be realised. Activating dihydrogen, for example, could be the first step in a hydrogenation process to catalytically convert an alkene into an alkane by adding dihydrogen across the carbon–carbon double bond. In a hydrogenation reaction catalysed by transition metals, the step after oxidative addition of dihydrogen would be the coordination of the alkene substrate, to which the hydrogen atoms transfer, before that cleaves off to regenerate the original metal centre and the process begins again.
But to get such a cycle to work, several traps must be avoided. ‘For the low coordination number main group compounds, if you activate the dihydrogen but the formed element–hydrogen bond is too strong, or if there is no more vacant coordination site for the second substrate, then this is a dead end,’ says Driess. Navigating through this tricky territory to put all the steps in place for a full catalytic cycle remains work in progress.
‘It is still being shown that low oxidation state p-block compounds can participate in the kind of reactions usually only associated with transition metals,’ adds Jones. ‘Real catalysis is yet to be achieved but I think certainly do-able, and within the next few years we’ll see some true catalysis coming out, especially from the group 14 elements silicon and germanium.’
Driess is even more bullish about the area’s prospects: ‘I would expect that in another 10 years, low valent main group compounds will be as good as traditional transition metal compounds for activating small molecules.’ As a researcher also known for his ground-breaking transition metal catalysis research, he is certainly well placed to judge.
James Mitchell Crow is a science writer based in Melbourne, Australia
The FLP side
The area of main group chemistry closest to being ‘marketable’ for use in an industrial situation for organic synthesis is that of frustrated Lewis pairs (FLPs), says Jones.

FLPs were a discovery made by Doug Stephan and his team at University of Toronto in Canada in 2006.7 ‘We were looking at titanium-based olefin polymerisation catalysts, using boron reagents as activators, stabilising some very active polymerisation catalysts with phosphine donors,’ Stephan recalls. ‘Going through a series of reactions, we discovered that very bulky phosphines did something quite different – they didn’t react with titanium, but just with the boron centre.’
Stephan discovered that he had made a particularly unlikely-sounding compound: one that would react with dihydrogen to incorporate both a proton (H+) and a hydride ion (H–), the former located at the phosphorus atom and the latter at the boron. This kind of compound sounds exactly like it should spontaneously lose dihydrogen, not take it up. Key to this behaviour is the use of bulky ligands in the parent boron and phosphorus compounds, thereby preventing the proton and hydride ion from recombining to form dihydrogen at room temperature. ‘Once you’ve activated hydrogen, the obvious next step is to use it in hydrogenation catalysis, so that’s what we did over the last few years,’ Stephan says. The team has successfully hydrogenated imines and alkenes in this way, and has now moved on to catalytic reactions involving activating carbon dioxide, a feedstock in no short supply.
‘Our first attempt was simply to capture carbon dioxide with FLPs. Then we found that if we moved to stoichiometric aluminium phosphorus we could get stoichiometric reduction to go, reducing carbon dioxide to carbon monoxide, and also carbon dioxide to methanol. Then the next evolution was to try to get a catalytic system,’ Stephan says. The process worked, but generated a troublesome by-product, phosphine oxide, in the process. ‘We need another oxygen acceptor that would be more valuable than phosphine oxide,’ Stephan summarises. ‘That work’s ongoing, but the good news is we can do catalysis without using precious metals.’











No comments yet