There's more than one way to engineer an enzyme, explains Kira Weissman.
There’s more than one way to engineer an enzyme, explains Kira Weissman.
Enzymes are extraordinary chemists. These protein catalysts accomplish impressive feats of chemical transformation in cells under very mild temperatures and pressures , all the while generating very little waste. Critically for us, they accelerate the rates of biological reactions (in some cases by as much as 1017 times) that would be otherwise too slow to support life. Perhaps more amazingly, their high efficiency does not hamper their ability to faithfully select one or a few target molecules from among the many in their environment, and to carry out very specific reactions on these substrates that would challenge even our most skilled synthetic chemists.
Understandably, there is enormous interest in co-opting enzymes to instead perform reactions of our choosing, outside of cells. Enzymes are already enjoying a great many practical applications: they are used in industry; in chemical synthesis; as reagents for biomedical research; in bioremediation; and, more recently, as therapeutics. However, there is a persistent barrier to exploiting them - these catalysts are specially predisposed to perform particular functions within living organisms and so are generally unsuited to other tasks.
Not only are enzymes fussy about which molecules they will accept and which reactions they will carry out, some are unhelpfully sluggish even with their natural substrates. In addition, they often dislike unfamiliar environments and may shut down completely when they encounter even small amounts of the products of their own reactions (a consequence of their participation in tightly controlled metabolic networks within cells). In contrast, biotechnology requires catalysts that tolerate just about any substrate, are functional and stable under a wide range of unnatural conditions, including organic solvents, and are active for long enough to generate lots of product. Even more desirable are enzymes that can carry out reactions never before seen in nature.
For the past few decades, researchers have tackled these limitations by attempting to ’engineer’ enzymes, changing their structures at the amino-acid level through appropriate DNA modification. Engineers usually set out to do one of the following: increase robustness and activity under a desirable set of conditions; introduce selectivity towards a new substrate; or, most ambitiously, elicit an entirely new function from an existing scaffold. Although nature has spent millions of years perfecting enzymes for their specific roles, it can only have sampled a tiny minority of protein sequences. For example, given that twenty different amino acids can occupy each position in a protein, the number of possible variants of even a small, 100-residue enzyme is 20100 - more than the number of atoms in the known universe! So, naively, it would seem that there is considerable scope for modifying enzymes to do our dirty work.
Most random sequences, however, will be devoid of a desired function and may even fail to fold properly. ’This sequence space [of a typical enzyme] is mostly empty - at least, mostly empty of the function you’re interested in.’ Explains Frances Arnold, professor of chemical engineering and biochemistry at Caltech, US. So, the most fruitful starting point for engineering is an enzyme that is already very similar to the desired catalyst in terms of its properties, reaction mechanism or substrate preference. The critical question, though, remains: where in the vast sequence-space of the existing enzyme to go looking for a new activity or enhanced features? Clearly, any laboratory-based exploration of this space can only reach a small subset of the possible sequences, and so must be carefully designed in order to be efficient. Since the mid-1990s engineers have been able to choose between two contrasting approaches for making this decision: ’rational design’ and ’directed evolution’.
The rational approach
Rational design means making an educated guess about which amino acids in an enzyme to alter, and then making the changes using targeted, or ’site-directed’, mutagenesis of the corresponding gene. This approach works best when a high-resolution, three-dimensional structure of the enzyme has already been obtained using X-ray crystallography or nuclear magnetic resonance (NMR) spectroscopy. Particularly helpful are structures in which the enzyme is ’caught in the act’ with its substrate, a substrate-like inhibitor or product. These structures help to identify the residues that are at the business centre of the enzyme - its ’active site’.
Even in the absence of a structure, it is still possible to create a computer model of a protein based upon the structure of enzymes that have similar sequences; a large database that correlates sequences with structures is readily available.
This information is used to predict (often computationally) the most promising sites in a protein to modify - generally one or at most a few amino acids. There is usually an obvious logic in the choice of residues: they are close to the active site and fit with the proposed reaction mechanism, are in the binding pocket for the substrate, or make important structural contributions to the enzyme. By changing the DNA that codes for these amino acids using standard molecular biological techniques, researchers can then substitute them with other residues.
Amino acids are usually exchanged with one of the other 19 natural residues, although it has recently become fashionable to also incorporate ’designer’ amino acids into proteins (see Chem. Br., July 2003, p24). The mutated gene is introduced into a suitable organism, which is then induced to make the variant protein. After purifying the modified enzyme away from all of the others in the cell, the researchers can then determine whether the amino acid substitutions have had the desired effect. For example, they might challenge the modified enzyme to work at temperatures that would simply cook the original. This process is typically iterative, with multiple rounds of mutation and evaluation.
Rational design is clearly information intensive, and a significant drawback to this approach is that the required structural and mechanistic data are available for only a tiny fraction of interesting catalysts. A much more significant limitation is that our understanding of enzymes lags far behind that of small molecules. Despite decades of intensive research, we don’t yet know how a protein’s sequence of amino acids dictates its final shape - the three-dimensional architecture that not only determines its function, but all of its other physical properties. More seriously, even if we could predict the structure, we wouldn’t be able to say what reactions the enzyme could perform or how well it could do them. Even if one trait is redesigned successfully (eg specificity), it is virtually impossible to predict the cost to another (eg stability) because even minor sequence changes may cause significant structural havoc in other parts of the protein.
Despite these challenges, rational design has been successful in an impressive number of cases. In the last couple of years alone, engineers have reconfigured the substrate specificity of multiple classes of enzymes, including oxidoreductases, hydrolases, transferases and DNA-cutting enzymes, in each case without changing the reaction mechanism. Rational design has also been used to alter co-factor requirements (eg from nicotinamide adenine dinucleotide phosphate (NADPH) to its significantly less expensive counterpart, NADH), invert reaction stereochemistry, and to enhance stability and rigidity under an assortment of unnatural conditions. It has even made inroads towards the ’Holy Grail’ of enzyme redesign: the introduction of a completely novel catalytic activity into an existing template.
Let the experiment decide
Notwithstanding its successes, for many researchers, rational design remains unacceptably hit-and-miss. So for the last ten years, they have been pioneering an alternative approach to engineering, called directed evolution (see Chem. Br., April 1999, p48). The technique is now flourishing in academia and a handful of biotech companies, such as Maxygen, Diversa, and Applied Molecular Evolution, have been founded to exploit it.
The selling point of directed evolution is that it does not require any understanding of the relationship between protein structure and function. Instead, it lets the experiment reveal the best solution to the engineering problem by replicating Darwinian natural selection in the laboratory. This ’perturbation-response algorithm’ has a proven track record in enzyme development, having successfully adapted natural catalysts to a staggering range of environments including volcanic acids, alkaline lakes, the arctic tundra and the ocean depths. Fortunately for us, in the lab it takes weeks instead of millions of years.
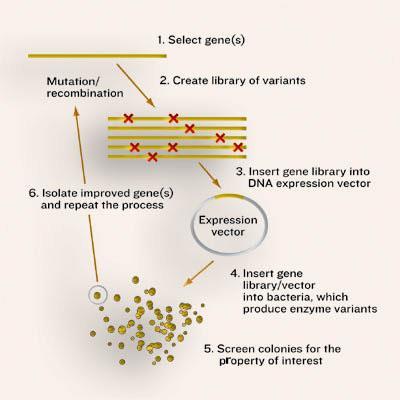
As in the natural process, directed evolution requires a large pool of enzymes with different sequences. In nature these variants result from random mutation of DNA, while in the lab genetic diversity is created deliberately. The larger the number of enzymes, the better the chance of finding one molecule with the desired behaviour. In nature, the ’winning’ enzyme, ie the one that makes it into the next generation, is that which confers the greatest survival benefit on its host - there is no particular direction in mind. In contrast, the lab version is decidedly goal-oriented. The challenge, though, is to select for a particular new trait or ability, many of which are not essential (or cannot be made essential) for an organism’s survival.
Getting random
The first step in any directed evolution experiment is to create very large numbers (typically tens of thousands) of gene sequences on which the selection regime can act. To this end, researchers have developed ways to make the normal process of duplicating DNA - polymerase chain reaction (PCR) - error prone so that, on average, each new copy of a gene contains a single, random mistake. Another strategy, developed by biochemist Pim Stemmer at Maxygen, introduces sequence variation along the entire length of each gene by recombining large chunks of DNA from multiple parents (gene sex, if you will). Originally this ’shuffling’ technique hinged on the similarity of the parent sequences, but newer methods (for example, the fancifully named ITCHY and RACHITT) allow sequences with much lower homologies to be mixed together, creating even more diversity.
In the next stage, the libraries of DNA sequences are introduced into microbial hosts (typically bacteria or yeast), so that each cell receives a slightly different version of the gene. The microbes contain all the essential supplies needed to produce proteins, and so the introduced genes are turned into enzymes within the cells. In this way, the enzyme and the instructions for its manufacture are ’packaged’ together.
The next step is to evaluate the mutant enzymes, one by one, for the greatest improvement in a desired property. The design of the evaluation stage is critical because it can only reveal what is screened for: ’your success depends on how well your screen measures what you really want,’ explains Arnold. In the rare cases that the cell’s survival can be made to depend on the enzyme’s activity (termed ’in vivo complementation’), selection of the catalytic victors is straightforward. More typically, the experimenter has to be able to analyse a large number of variants for particular traits without sacrificing the ability to measure even minor improvements. To this end, individual cells are usually grown in the wells of multi-well plates to form tiny cultures (as many as 1536 of them on each plate), whose properties can then be evaluated using a wide array of high-throughput analytical devices.
To date, most screening for activity has been based on turning substrate into a fluorescent or highly coloured product. So cells harbouring the most efficient enzymes fluoresce more intensely (or turn bright yellow) and so can be easily selected. As this approach is inherently limited to certain reactions, however, researchers in the field are concocting increasingly clever new screening methods that are in principle applicable to any type of chemistry. For example, in the US, Columbia University chemistry professor Virginia Cornish and colleagues have devised a system where the level of catalysis exhibited by an engineered enzyme is linked to the activation of a ’reporter’ gene, which in turn produces an obvious change in the cells’ appearance (such as turning them from blue to white). Their model system is already suitable for a range of enzymes that catalyse both bond-formation and bond-breakage, such as glycosyltransferases, aldolases, esterases, amidases and Diels-Alderases.
The final step in the process is to retrieve the winning genes from the selected cells and to use them as parents for the next generation of potential catalysts. The mutation/selection process is then iterated in the hope that beneficial mutations begin to accumulate. In the best cases, the result is a new enzyme that is highly evolved for a particular function or attribute. Interestingly, sequencing of the resulting mutants often reveals changes that would never have been predicted ’rationally’ using the 3D structure of the protein - further validating this ’random’ approach to enzyme engineering.
Over the last decade, directed evolution has proved that it can compete with the real thing, as designed by Mother Nature herself. Since 2001 alone it has been used to enhance enzyme properties such as thermostability, tolerance to organic solvents and pH profile. Catalysts have successfully been developed for detoxification and bioremediation (eg degradation of organophosphates and chlorinated aliphatics) and novel specificities and activities have been introduced into enzymes. The technology has made it possible to change reaction stereospecificity, and even to evolve whole systems of enzymes. However, like rational design, it has as yet failed to elicit fundamentally different chemistry from an existing active site.
The way forward
While enzyme engineers typically fall into either the ’design’ or ’evolution’ camp, it is increasingly clear that a combination of approaches is the best way forward. As our understanding of structure-function relationships improve, it should be possible to launch a more focused attack on particular regions of the protein landscape, and so optimise evolution experiments. In the end, it may be a ’rationally random’ approach that finally enables engineers to create novel enzymes that are able to perform any reaction we desire.
Acknowledgements
Kira Weissman is a Royal Society Dorothy Hodgkin Fellow in the department of biochemistry, University of Cambridge, UK
Further Reading
H Tao and V W Cornish, Curr. Opin. Chem. Biol., 2002, 6, 858.
D N Bolon, C A Voigt and S L Mayo, Curr. Opin. Chem. Biol., 2002, 6, 125.
T M Penning and J M Jez. Chem. Rev., 2001, 101, 3027.
U T Bornscheuer and M Pohl, Curr. Opin. Chem. Biol., 2001, 5, 137.






No comments yet