Researchers in the UK have developed a transformer-based model for quickly predicting the physical properties of molecular crystals. Called Molecular Crystal Representation from Transformers, or MCRT for short, the team behind the model liken it to ChatGPT. Andy Cooper, from the University of Liverpool, who was part of the team, says their ‘“Crystal GPT” [was] trained on the largest available database of organic crystals,’ and ‘has learned the most distinctive patterns within these crystals, using multiple representations such as symmetry and crystal density, and how these patterns relate to practical properties’.
Understanding and predicting crystals’ properties is a key element of materials design, but doing so through traditional computational methods is often resource-intensive. Machine learning has emerged as a promising alternative, offering faster and more cost-effective predictions of structure–property relationships. Techniques such as Smooth overlap of atomic positions (SOAP) and atom-centred symmetry functions (ACSFs) can effectively predict properties such as lattice energy, while geometric descriptors – including accessible surface area and pore diameter – are useful for capturing global material characteristics. However, these methods come with limitations. SOAP calculations are memory-intensive, and geometric descriptors tend to compress structural information, potentially overlooking subtle but important details. Moreover, ‘state-of-the-art deep learning approaches are typically problem specific and cumbersome to re-train for new tasks or data,’ explains team member Xenophon Evangelopoulos, also at the University of Liverpool.
Evangelopoulos says ‘MCRT was intended to be a foundation model that can be easily fine-tuned to the problem at hand, even with small amounts of available data.’ While Graeme Day from the University of Southampton, who was also part of the MCRT team, says their model won’t necessarily replace other methods of crystal structure prediction, ‘however, it might play a role in improving or speeding up the stability ranking of existing methods’.
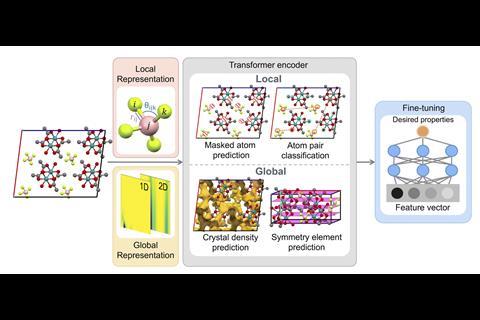
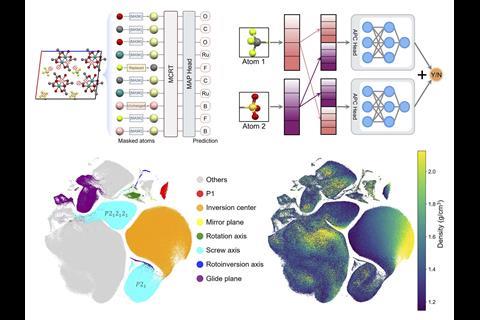
Minggao Feng, another team member at the University of Liverpool, explains that they pre-trained MCRT on 706,126 experimental crystal structures extracted from the Cambridge Structural Database (CSD) ‘using tasks designed to enable the model to learn important patterns about both the crystal’s local atomic environments, and its overall global geometry’. Feng says the model architecture uses a multi-modal blend of features, comprising graph-based atomic representations and topological “images” that help build a rich universal understanding of the crystalline nature of the material. ‘This produced a foundational model that we can then fine-tune – that is, re-train – to any specific family of crystals and any desired functionality. The model’s attention-based architecture also offers some explainability features that can be used to “reverse-engineer” predictions and highlight which structural features contributed most to the prediction, making these predictions less “black box” in nature.’
Day says ‘MCRT’s major benefit arises in low data availability cases where it can demonstrate impressive performance even in very-few-shot learning scenarios.’ ‘Chemistry laboratory experiments, and some calculations, are expensive. There is huge power in being able to extrapolate reliably from a small number of observations, and transformer models are one way to do this,’ adds Cooper.
Keith Butler, who leads the materials design and informatics group at University College London in the UK, explains that predicting properties from small datasets is difficult because machine learning models typically require large amounts of training data, which are often unavailable in this field. ‘It’s cheap to get labels of pictures of cats or dogs or street scenes, but getting labels of how methane diffuses in a material is very different, you can’t just use a captcha to scrape that kind of high-value data! So what is great is that by training their models on similar tasks, where they do have lots of data, they can then fine tune them for these tasks where there is not lots of data and the model works really well despite the limited training set. This opens up lots of new possibilities for computational design of new functional organic crystals.’
‘This work marks a transformative step toward AI-driven materials chemistry,’ comments Michael Ruggiero from the University of Rochester in the US, whose research on atomic motion in the condensed phase saw him included in Forbes’ 30 under 30 list in 2019. ‘By combining rigorous physics-based insights with a universal, transferable architecture, the MCRT model opens the door to faster, more cost-effective prediction of molecular crystal properties – redefining how we approach materials design in silico and accelerating the pace of materials discovery. This work will revolutionise the way in which modern chemistry research is performed, and will ultimately enable the ability to experimentally realise new materials for advanced applications.’
MCRT was pre-trained on four tasks:
- Masked atom prediction. This gives the model a deeper understanding of the local chemical environment.
- Atom pair classification. This identifies whether a pair of atoms are from the same molecule, which is designed to help the model distinguish the different molecules within a crystal cell, and to improve understanding of the crystal structure.
- Crystal density prediction. This gives important global information about the crystal, particularly for predicting properties which rely on crystal voids and porosity.
- Symmetry element prediction. This gives information about the crystal space group, based on the crystal symmetry elements present.
References
This article is open access
M Feng et al, Chem. Sci., 2025, 16, 12844 (DOI: 10.1039/d5sc00677e)









No comments yet