

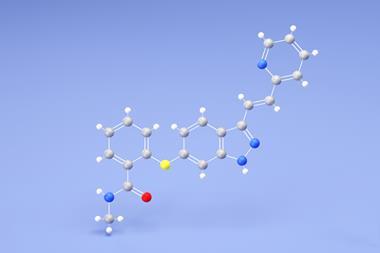
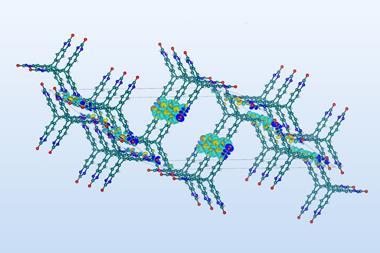
Getting crystal structure predictions right can save pharmaceutical companies hundreds of millions of dollars. Yet while the pharmaceutical industry might have a keen interest in predicting what crystal structures can form, and how stable they are, computational costs have priced out calculating the relevant properties for each compound from first principles. Now, Marcus Neumann, founder and chief executive of Avant Garde Materials Simulation (AMS), a crystal structure prediction software company, alongside colleagues from academia and industry have demonstrated the ability to calculate crystal structure stability as a function of temperature and humidity, as well as indicating how accurate the calculations are expected to be for different compounds.
‘There was this kind of gold-rush feeling about crystal structure prediction in the community, related to the fascinating phenomenon of disappearing polymorphs and the potential commercial value of late-appearing crystal forms,’ explains Neumann. A case in point was the antiretroviral drug ritonavir used to treat HIV/Aids, as well as hepatitis C and Covid-19. Although now included in the World Health Organization’s List of Essential Medicines, in 1998 just two years after it was introduced ritonavir was temporarily removed from the market altogether when a slower nucleating but more stable crystal structure with much lower therapeutic effects was discovered and found to dominate supplies.
The stability of a crystal structure is determined by a thermodynamic quantity known as the ‘free energy’. However due to the difficulties in calculating the free energy of a crystal structure, for a long time people calculated crystal lattice energies at the ‘classical zero-Kelvin limit’ instead. This assumes the structure is infinitely cold and bereft of quantum effects. In reality, both quantum effects and the atomic motions observed at finite temperatures contribute substantially to the lattice free energy, and it is essential to calculate these energies correctly to establish the effects of temperature and humidity on the relative stability of crystal forms, a potential problem for pharmaceutical companies. ‘If you have a lab in the UK the humidity will not be the same as in Hong Kong,’ explains Elsa Gheziel, who shares the role of chief executive at AMS with Neumann. ‘You do not know what impact it will have on the crystallisation behaviour.’
Unique position
Efforts to extract more realistic values were kickstarted by study co-author Alexander Tkachenko with Jonannes Hoya, who demonstrated their approach in the 6th blind test on crystal structure prediction in 2016. Since then, Neumann and colleagues have been finessing the approach to be more robust and faster. They have been developing Grace – the Generation, Ranking and Characterisation Engine for crystal structure prediction. Their latest advance concerns the final stage in the crystal structure prediction workflow – the calculation of reliable lattice free energies.
Access to experimental data from multiple pharmaceutical partners also placed the team in a unique position for quantifying what level of trust they could put into their crystal structure predictions. ‘No one in the world has 37 reliable free energy differences for molecular crystals,’ says Neumann. By comparing their calculated values against the empirical data, they quantify error bars for their theoretical predictions, from which they then extrapolate the error per water molecule and per non-water atom to give a steer on the program’s accuracy for compounds not yet encountered.
Matteo Salvalaglio at University College London – who was not involved in this research – describes the computational prediction of the most likely crystal form of organic molecules as ‘a formidable task’. He adds that the work ‘redefines the boundaries of the state of the art in the prediction of thermodynamic properties of crystalline materials’, and approaches ‘real world applications’.
However, while Neumann describes crystal structure prediction as now ‘more or less a solved problem’, Salvalaglio suggests that ‘predicting the crystal structure of organic compounds is a broader question than the one addressed by an accurate thermodynamics determination’ and that ‘predicting the likelihood of observing polymorphs, and the conditions in which they can be obtained remains an open research question’. In fact Neumann is keen to tackle the design of methods for observing the many experimentally elusive crystal structures predicted in silicon next. If forms are predicted to be more stable than those experimentally observed, crystallising them is essential to remove the risk of their appearance and dominance at a later date, as in the case of ritonavir. Crystallising less stable forms could also benefit drug development because of their increased solubility and hence bioavailability. In addition, Neumann would like to develop ways of helping drug companies navigate decisions over the choice of salts, solvates and co-crystals – the solid-state forms – that will ‘keep them out of trouble’ in terms of bioavailabilty, processability, shelf-life and the risk of more stable forms making a later appearance.
References
D Firaha et al, Nature, 2023, DOI: 10.1038/s41586-023-06587-3












No comments yet