
A software package for automatic conformational analysis can now compute the entropies of flexible molecules with little user intervention.1 This new feature builds upon the program’s ability to map out conformer landscapes, and exploits a balance of accurate calculations and insightful approximations.
Changes in entropy can make a significant contribution to the thermodynamics of a reaction, particularly if a molecule becomes bound or loses its flexibility. ‘An automated protocol to calculate it provides a good starting point for further quantum and experimental chemical investigations,’ says Cristina Trujillo, a computational organic chemist at Trinity College Dublin in Ireland, who was not involved in the research. Predicting a molecule’s entropy depends on knowing the states a molecule can be found in and their relative energies, such as rotational and vibrational energy levels. For flexible molecules like alkanes, the distribution of molecules between low-energy conformers is a significant additional source of entropy, and the conformers have differing rovibrational entropies. This makes calculating the overall entropy for these molecules challenging.
The Crest software package, developed at the University of Bonn in Germany, can already find conformers by automatically designing and running metadynamics searches, which solves part of the puzzle. ‘Prior to Crest there weren’t really straightforward ways to calculate the conformational entropy, only a few specialised tools in a few groups’, says Geoffrey Hutchison of the University of Pittsburgh in the US, who recently used Crest in a molecular entropy study.2 Although finding conformers is simplified by Crest, calculating the entropy is still a nuanced process which places strict demands on the quality of the search results. ‘Everything has to come together,’ says Stefan Grimme, Crest’s lead developer. ‘You need a good method for the potential energy surface, It must be robust, reasonably fast, it has to be general, you have to have the algorithms, and you have to really make it quantitative not just for molecules that are rigid or semi-rigid but for really very flexible ones.’
It’s not just something for the expert
Nevertheless, automatic entropy calculations were a natural next step in the program’s development. ‘Even in the oldest version we had a printout of a rough estimate of the conformational entropy,’ Grimme notes. In some respects, Crest had a head start: for example, its NMR functionality allowed it to recognise distinguishable and indistinguishable rotamers, which is essential to properly counting the available states.
Extrapolate to completeness
The entropy calculation starts with a version of Crest’s search algorithm, which is modified to emphasise completing the conformer ensemble. Crest monitors the improvement of an estimated conformational entropy as more conformers are found. ‘One question in conformer searching in general is “do we have the full ensemble”, and the new workflow outlines a nice criterion,’ says Hutchison. ‘Over time, there’s a convergence to the exact entropy.’ Because of this convergence, an accurate conformational entropy can be extrapolated from a thorough, but finite, conformer search. ‘Without this extrapolation of the entropy, you cannot make it efficient,’ notes Grimme. At this stage the most important conformers have been identified, and their rovibrational entropies can be calculated. The ensemble average of the rovibrational entropies, added to the extrapolated conformational entropy, is the total molecular entropy.
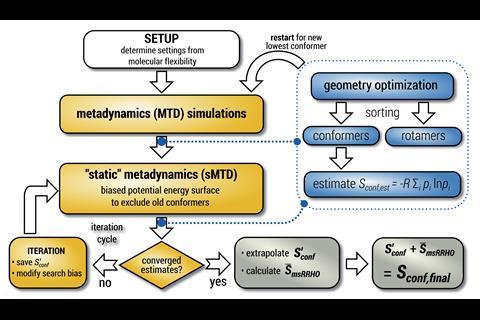
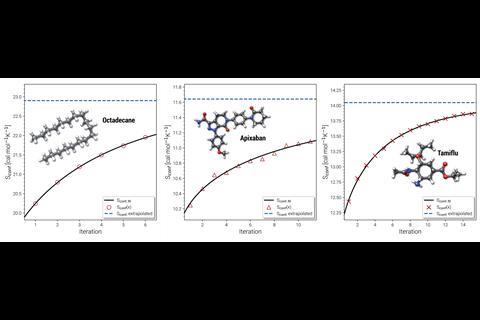
The new function builds upon the Crest’s foundations to good effect: for a test set of 62 gas phase molecules, Crest’s predictions match experiment to within about 1–2cal/mol K. A closely related quantity, the heat capacity, can also be computed with similar accuracy. ‘I think that we have a method where you can press the button and then you really get a value that is very close to experiment,’ says Grimme. ‘It’s not just something for the expert.’
Trujillo is cautious. ‘In my case, we work with organocatalysts with more than 100 atoms,’ she notes, versus the 98 atom maximum in Grimme’s benchmark set, and Crest has not been entirely reliable at predicting conformers in her studies. She would like to see the method proven with a wider variety of problems, particularly with solvent models. ‘Solvent is compulsory in our research in order to mimic experimental conditions, and especially when non-covalent interactions are involved.’ However, Hutchison is pleased with how Crest’s results compare to his own calculations. ‘We’ve done some initial benchmarking and they seem quite similar. If you want fully accurate thermochemistry, you absolutely want to use the new workflow.’
References
1 P Pracht and SGrimme, Chem. Sci., 2021, DOI: 10.1039/d1sc00621e (This article is open access.)
2 L Chan, GM Morris and GR Hutchison, J. Chem. Theory Comput. 2021, DOI: 10.1021/acs.jctc.0c01213












No comments yet