
Quantum chemical calculations drive program that quickly and easily builds up plausible ensembles of molecular conformers
A new program has been developed to suggest the conformations adopted by molecules as they fold and pivot. The program combines a high degree of automation, modest computational demands and broad chemical flexibility, and has already been adopted successfully by experimental and theoretical chemists.
Look at a diagram of a molecule and it’s easy to imagine that it’s a static object. However, chemical bonds can rotate, so in reality molecules flop and fold and take different shapes, called conformers. Because the properties of conformers can differ, researchers often need to know what a molecule’s possible conformers are. That could mean identifying a specific conformer, or averaging the behaviour of countless conformers over the timescale of an experiment, or anything in between.
Although identifying relevant conformers is an important problem in chemistry, there are few tools to automate it, leaving researchers to devise their own solutions or rules of thumb. Trying to manually but exhaustively explore the options is time consuming and prone to error, notes Yunjie Xu, an experimentalist studying chiral molecules at the University of Alberta in Canada. ‘We basically look at molecules and decide, “what are the rotatable bonds?”’ she says. ‘We would do the rotation for every single one of them, and very quickly it became too tedious.’
Then one of Xu’s postdocs introduced her to new software from the theoretical chemistry research group of Stefan Grimme at the University of Bonn in Germany. Their conformer–rotamer ensemble sampling tool (Crest) program automatically searches for conformers of a molecule, discarding duplicates as it goes. It continues until it has discovered the set that is lowest in energy, and therefore most experimentally relevant, within constraints given by the user. The code has been designed to be easy to use, computationally efficient, and relevant to many different chemical problems.
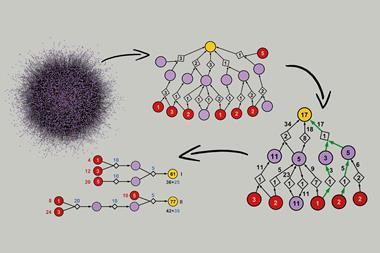
Iterative metadynamics search
Crest uses a technique called metadynamics. Chemists can identify low-energy conformers by simulating a molecule’s flapping conformational dance, a technique called molecular dynamics, and noting down the conformers they see. However, the few lowest-energy conformers would dominate the observations. Metadynamics applies fictitious forces to the atoms that push the molecule away from conformations it has already taken, so it finds new ones at slightly higher energies. Metadynamics is already used to search for conformers, but designing the simulations is a nuanced process.
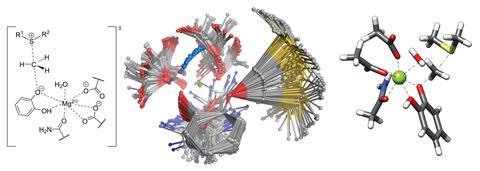
Crest performs an iterative metadynamics search for the most important conformers of a molecule, automatically. The cycle begins with a set of metadynamics calculations that begin with a specific conformer, but have a wide range of simulation parameters. Those searches expose a variety of low-energy conformers, which are subjected to gentler molecular dynamics searches. The best conformers found up to this point are spliced with each other to introduce some novel candidates, and the structures and energies are refined and recorded. The code then starts another cycle from the current lowest-energy conformer. Crest continues in this fashion, attempting to find an even-lower-energy set of conformers, until the lowest-energy conformer doesn’t change with each cycle.
This is an unusual theoretical group that is very aware of experimental data
This approach is unbiased and indifferent to the molecule being studied. ‘The metadynamics machinery in Crest works with the Cartesian coordinate system and a rather general biasing potential,’ Grimme explains. ‘This makes it general and [it] requires no specific user input.’ Because the molecules are described with efficient models developed in Grimme’s group, ‘it is accurate, robust, and still fast enough so that it does not require any super computer resources, but basically a laptop only’. ‘Crest provides a good balance between accuracy and computational cost,’ comments Cristina Trujillo, whose research at Trinity College Dublin in Ireland involves computational studies on reaction mechanisms. ‘Conformational search is usually the first calculation [we do]. In this context, this methodology is a very useful tool.’
Conformers aren’t the only variations on a molecule that chemists might be interested in, so Crest includes a selection of extra functions. The code notes rotamers – conformers where equivalent atoms are exchanged – as these are important in NMR. Some molecules can protonate in varied ways, so Crest can generate tautomers and protomers. It can even predict different structures of small molecular clusters. The software can already search for conformers of transition states, for reactions in those clusters, but future developments may allow it to explore reactivity in a cluster more generally, making what Grimme calls a nanoreactor.
Xu credits the program’s success to an awareness of experimentalists’ needs: ‘This is an unusual theoretical group that is very aware of experimental data, and understands what data they’ll use to compare it with, and what the experimental data means, and that’s not that common.’
References
This article is free to access until 15 April 2020
P Pracht, F Bohle and S Grimme, Phys. Chem. Chem. Phys., 2020, DOI: 10.1039/c9cp06869d












No comments yet