Understanding machine learning predictions by exploring the road not travelled
Machine learning methods can efficiently solve complex problems, by training models on known data and applying those models to related problems. However understanding why a model returns a particular result, which is vital to validating and applying this information, is often technically challenging, conceptually difficult and model-specific. Now, a team in the US working on explainable AI for chemistry has developed a method that generates counterfactual molecules as explanations, which works flexibly across different machine learning models.1
‘There’ve been some high profile accidents in computer science where a model could predict things quite well, but the predictions weren’t based on anything meaningful,’ explains Andrew White from the University of Rochester, whose team developed the new counterfactual explanations method. ‘Sometimes [a machine vision model] will predict that there’s a horse in a picture, not because there’s a horse in the picture, but because there’s a photographer’s watermark. Missing a picture of a horse is obviously low stakes, but if you try to predict if something is carcinogenic or toxic or flammable, then we start running into problems that are more serious.’ Understanding whether a model has reached the right answer for the wrong reasons – known as the Clever Hans effect after a mathematically-gifted horse – is one of the goals of explainable AI.2
Counterfactuals are an intuitive and informative explainable AI approach. For any particular prediction, for example that an input molecule is soluble, a counterfactual is the most similar example where the model gives a different prediction. ‘Through a comparison of what has changed, for example the loss of a carboxylic group resulting in a change of chemical activity, you “learn” why the model is giving the prediction it is,’ explains Kim Jelfs, a researcher in computational materials discovery at Imperial College London, UK. ‘This is intrinsically rather a satisfying way for a chemist to understand how a machine learning model is working.’ If the model is behaving well, this counterfactual is also a useful prediction in its own right. ‘A counterfactual explanation is actionable, it tells you how to change your molecule to change its behaviour,’ notes White. ‘It’s giving you a real molecule that you could synthesise and test.’
A counterfactual explanation is actionable
However, searching for a counterfactual still typically depends on the subtleties of the specific AI model being used. ‘Let’s say you’re working with a graph neural network,’ says Geemi Wellawatte, a researcher on White’s team. ‘You need special attention because you’re working with the graph rather than a string representation [of a molecule]. Most of these explainable AI methods have been very sensitive to the model, and the downside is that your method cannot be applied in general, no matter how good it is.’ Searching for a most-similar molecule is also uniquely challenging. ‘Taking the derivative with respect to molecular structure is a very odd kind of concept and it’s numerically very difficult,’ explains White.
Leave no stone unturned
The answer was to use a simpler method to make similar molecules. White’s student Aditi Seshadri suggested they try Stoned, the Superfast traversal, optimisation, novelty, exploration and discovery method developed at the University of Toronto in Canada, which generates a molecule’s chemical neighbours by modifying the Selfies string that describes it.3 ‘This is such a simple method to use: no derivatives, no GPU, no deep learning. It’s just literally modifying strings,’ White enthuses.
This idea led the team to create Mmace – short for Molecular model agnostic counterfactual explanation. Mmace takes a molecule and uses a fine-tuned Stoned search to build a library of similar molecules. These can be screened with the machine learning model to see which molecules give different results, and the Tanimoto distance shows which are most similar.
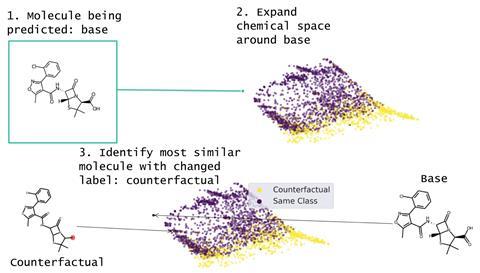
As Mmace doesn’t depend on the internal structure of the machine learning model, it’s simple to implement and broadly applicable. ‘A lot of times in machine learning research, researchers may prefer to change the model they are using depending on data availability or on the specific property being predicted,’ explains Heather Kulik, a chemical engineer at the Massachusetts Institute of Technology in the US who studies machine learning in chemistry. ‘Having an approach for model interpretability that applies to multiple types of machine-learning models will ensure its broad applicability.’ Jelfs is also pleased by the convenience of Mmace. ‘As they provide their approach open-source, others can immediately use it for interpreting their own deep learning models. Their method can be applied to any machine learning model, so it’s immediately very usable in the community.’
White’s team tested Mmace across a wide variety of chemical problems and machine learning models, including HIV activity prediction with a graph convolutional network, and solubility with a recurrent neural network, in each case obtaining counterfactuals which helped justify the properties of the original molecule. ‘How do you demonstrate that you’ve succeeded?’ wonders White. ‘We tried to look at this from a lot of different angles, but at the end of the day, “what is a valid explanation?” is such a nebulous concept that we were working it out with a philosopher.’
It’s immediately very usable in the community
White is keen to stress that Mmace isn’t a panacea. Selfies has difficulty representing some classes of molecules and bonding such as organometallic structures like ferrocene, and although all Selfies strings meet some criteria for chemical sense – atomic valence, for example – not all Selfies structures are necessarily synthesisable molecules. To address the latter, White’s team tried a similarity search on the PubChem database of experimentally reported molecules to generate chemical neighbours, instead of Stoned. This gave counterfactuals that were more different to the original molecule, but which still provided useful insights: modifications on a tertiary amine in a molecule removed its predicted ability to permeate the blood–brain barrier, implying that group has a role in allowing the molecule to cross.
White and his team are continuing to work on the nuances of the method, such as their definition of molecular similarity. ‘Maybe an organic chemist would think, “I could just synthesise this one with this pathway, and then if I make one little change, I could synthesise this one, and so they may be one step apart,”’ White explains. ‘We’re also creating explanations with the same tools, but trying to categorise these similar molecules into mechanistic explanations. We like the idea of communicating in only chemical structures for explanations, like counterfactuals, but at some point we need to align the explanations with our mental models of why a molecule works or does not.’
References
These articles are open access
1 G P Wellawatte, A Seshadri and A D White, Chem. Sci., 2022, DOI: 10.1039/d1sc05259d
2 S Lapuschkin et al, Nat. Commun., 2019, 10, 1096 (DOI: 10.1038/s41467-019-08987-4)
3 A Nigam et al, Chem. Sci., 2021, 12, 7079 (DOI: 10.1039/d1sc00231g)












No comments yet