Ever struggled to assign your NMR spectra? Can’t tell which diastereomers you have made? Help is here. Researchers in the UK have developed a program that automatically analyses 1H and 13C NMR data, assigns the peaks, and suggests which structure is most likely from a set of candidates.
Trying to work out what structure you have can be tricky, especially for if there are regioisomers or diastereoisomers with subtle differences in 1D NMR. One way to solve this is to use 2D NMR techniques, but these can be time consuming so it can be quicker to use computational methods instead. Building on an open source program called DP4, which predicts how likely it is that a candidate structure is correct by comparing predicted spectra with experimental spectra, Jonathan Goodman and his University of Cambridge co-workers have now developed DP4-AI. Goodman says DP4-AI ‘affords fully automated resolution of structural uncertainty, saving time interpreting NMR spectra whilst simultaneously giving confidence in the analysis.’
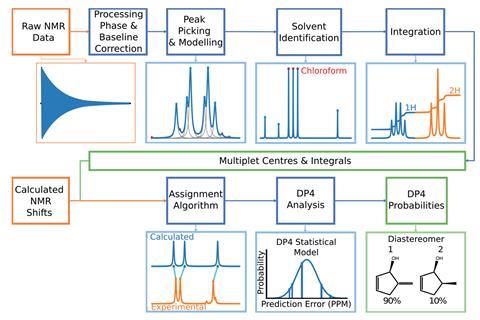
Standard DP4 requires the user to input their experimental NMR data: peak locations and a description of which atoms in the candidate molecule are chemically equivalent. The aim of DP4-AI is to remove this human input. It can take the raw NMR data and automatically process, analyse and compare the spectrum to DFT-calculated spectra to output a quantitative measure of confidence in trial structures. DP4-AI can do the full calculation for a molecule in about 60 seconds, compared to the manual process that could take up to 8 hours of a user’s time. This is not the same as the commercial software Mnova. Mnova aims to help users process and interpret their spectra, while DP4-AI uses an assignment routine coupled with DFT calculations.
‘Goodman’s group has pioneered the development of useful toolboxes to facilitate structural and stereochemical assignment,’ says Ariel Sarotti at the National University of Rosario in Argentina who works on organic synthesis and uses computational methods to study structure, selectivity, and reactivity. ‘Considering that the method is available as open-source software, I think it will be a popular approach in the near future.’
‘A number of challenges have made DP4-AI a difficult project to work on,’ explains Goodman. One major challenge was getting a set of molecules with corresponding raw NMR data that was publically available to demonstrate how well their method worked with real world examples. ‘Our quest for data has raised questions about the suitability of how NMR data is currently stored,’ says Goodman. ‘Although many groups keep raw NMR data, the required labels and corresponding structures are often scribbled in dusty lab books that have been confined to a shelf. Even if the data is accessible, its meaning may quickly be lost. This will affect the reliability and how repeatable organic chemistry is.’
Goodman is not concerned that the advance of automated processes will reduce the ability of organic chemists to analyse NMR spectra by hand. ‘Calculators have not stopped people doing arithmetic, but rather have allowed people to perform complex arithmetic more quickly and accurately.’ He plans to explore how they might extend DP4-AI to other nuclei, not just 1H and 13C. ‘We are excited by the ideas that the chemistry community has for DP4-A. Anyone can use and edit the system in their own way to solve interesting questions (and people already are). We will be very interested to see how people use and develop this software in the future.’
References
This article is open access
A Howarth, K Ermanis and J M Goodman, Chem. Sci., 2020, DOI: 10.1039/d0sc00442a









2 readers' comments