When it comes to cascade reactions, radicals are king of the ring-formers
Radical chemistry – despite its unique advantages – is often an afterthought for many practising organic chemists. In fact, the scope of radical chemistry has been consistently underestimated ever since its inception in 1898. Its discoverer, Moses Gomburg, attempted to reserve the entire field for his own personal study. The health industry, with its aggressive marketing of antioxidants, appears to live in constant fear of free radicals, and many inexperienced organic chemists are no different.
Unlike other common reactive intermediates, such as carbocations and carbanions, free radicals are neutral. That means their chemistry is no longer readily explained using familiar undergraduate terms like ‘electrophile’ and ‘nucleophile’. But the reactivity of many radicals – particularly carbon-centered radicals – does follow a predictable set of rules, and these neutral species are often a lot more selective than their charged counterparts.
One well-known facet of radical chemistry, even to those with little direct experience in the field, is its unique aptitude for cascade processes. These can rapidly form multiple bonds and rings in a single step. It’s no coincidence that the world record of forming seven rings in a single chemical reaction was achieved using a radical cascade.1 These reactions are so efficient that, during the 1960s, several high-profile chemists argued persuasively that this is surely how nature assembles the complex tetracyclic steroid ring system. We now know that steroid synthesis – along with the vast majority of nature’s biosynthetic cascades – is cationic rather than radical-driven. In the laboratory, however, without the benefit of enzymes to protect the highly reactive intermediates found in cationic reactions, radical polycyclisations are usually preferred.
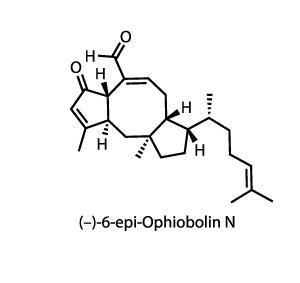
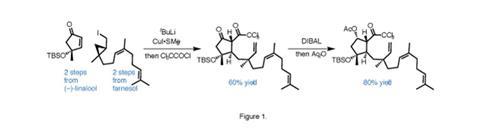
The power of radical cascades is demonstrated again in the seemingly effortless conquest of (–)-6-epi-ophiobolin N by Thomas Maimone and co-workers at the University of California in Berkeley, US.2 The group’s synthesis starts with an interesting three-component coupling between an iodocyclopropane, an enone and an acyl chloride. Lithium–halogen exchange on the iodide forms an alkyllithium that then rearranges, opening the cyclopropane. Copper-catalysed addition to enone, trapping the enolate with trichloroacetyl chloride, followed by reduction and protection of the ketone, gives the cascade precursor with blinding speed (figure 1). One common drawback to using cascade reactions is that the elaborate starting materials for these sequences can take more time to prepare than the cascade itself saves – the long way round to a shortcut, so to speak – but in this case the team assembles the starting material in short order.
One interesting detail is the group’s choice of starting materials. The two building blocks are prepared from linalool and farnesol, which are themselves terpenes. There’s something aesthetically appealing about building up a sesterterpene (with 25 carbon atoms) from a diterpene (10 carbons) and a sesquiterpene (15 carbons), mirroring nature’s routes to these molecules.
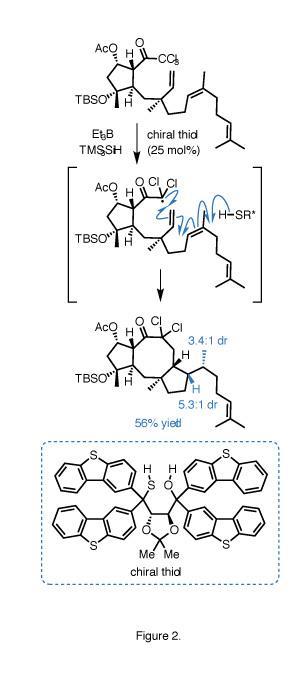
The cascade itself (figure 2) begins with reductive radical generation from the trichloromethyl group using TMS3SiH and triethylborane (a less toxic but more expensive alternative to the classic pairing of Bu3SnH and 2,2’-azoisobutyronitrile [AIBN]). This radical adds to the nearby vinyl group, closing the molecule’s intimidating central cyclooctane ring, before cyclising further onto the geranyl tail, and finally picking up a hydrogen atom to terminate the sequence.
For this final step, a chiral hydrogen-atom transfer catalyst is crucial to obtaining the necessary diastereoselectivity at the newly formed methyl stereocentre. With an achiral thiol, substrate control gives the unwanted epimer. Fortunately a large Taddol-derived thiol can override this innate preference to deliver the desired product.
With the tricyclic cascade product in hand, a handful more steps complete an elegant and concise synthesis.
BRSM is a process chemist and blogger based in the US
References
1 S Handa, P S Nair and G Pattenden, Helv. Chim. Acta, 2000, 83, 2629 (DOI: 10.1002/1522-2675(20000906)83:9<2629::AID-HLCA2629>3.0.CO;2-W)
2 Z G Brill, H K Grover and T J Maimone, Science, 2016, 352, 1078 (DOI: 10.1126/science.aaf6742)












No comments yet