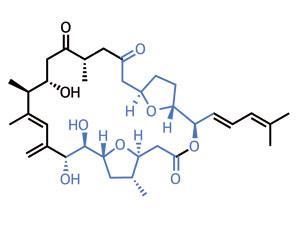
Again, we’re plunging into the marine depths to find natural products with prodigious biological activity. The amphidinolide family comprises over 30 members, varying in architecture but (almost) all featuring a complex and highly decorated macrolactone ring at the core. Amphidinolide F was first isolated in 1991, but as yet remains unconquered territory in synthetic laboratories.1,2 However, new ground has been broken by a pair of chemists from Oregon State University, US, led by Rich Carter.3 Their key insight was that hidden symmetry exists in the complex tetrahydrofuran (THF) regions. Although these two regions are not identical, the team considered that enough chemistry was in common that a mutual precursor might be used.
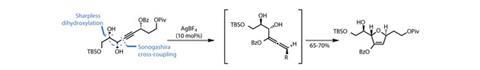
Eager to test their hypothesis, the team targeted the tetrahydrofuran precursor from the off, building a linear fragment based around an acetylene. The chains to either side of the acetylene are heavily oxygenated, with one set of hydroxyls installed using the ever-reliable Sharpless asymmetric dihydroxylation chemistry and the other half derived from malic acid. Treating the alkyne with a relatively strong Lewis acid prompted cyclisation of the nearer of the two unprotected hydroxyl groups onto an intermediate allene, neatly preparing the five-membered THF ring and avoiding a potential six-membered tetrahydropyran by-product (figure 1). Importantly, the team performed this chemistry repeatedly on multi-gram scale to supply their synthetic forays.
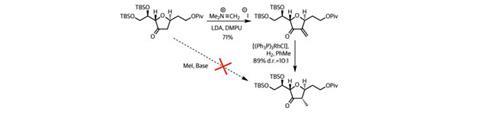
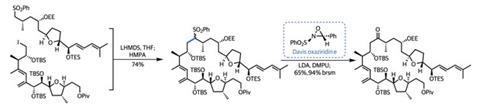
To bring about this reversal of reaction polarity, or umpolung, the chemists made a sulfone and deprotonated it with a strong base. This anion swiftly displaced the alkyl iodide to generate the required C–C bond in good yield. Of course, the team no longer required the sulfone group, so a little base, followed by the exotic-looking Davis oxaziridine provided the desired ketone (figure 3).4
Paul Docherty is a science writer and blogger based in Reading, UK
References
- J Kobayashi and T Kubota, J. Nat. Prod., 2007, 70, 451 (DOI: 10.1021/np0605844)
- J Kobayashi et al, J. Antibiot., 1991, 44, 1259 (DOI 10.7164/antibiotics.44.1259)
- S Mahapatra and R G Carter, Angew. Chem., Int. Ed., 2012, 51, 7948 (DOI: 10.1002/anie.201203935)
- J R Hwu, J. Org. Chem., 1983, 48, 4432 (DOI: 10.1021/jo00171a067)












No comments yet