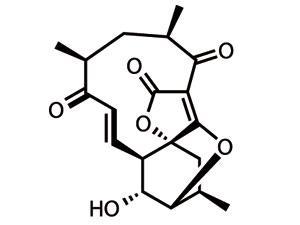
So that explains most of the target’s name, but not everything. This member of the abyssomicin family is the only one to achieve such bacteria-bashing prowess, and is also the only one to feature atropisomerism – a relatively unusual form of stereoisomerism in naturally occurring species. This structural feature is found when rotation about a single bond is sufficiently hindered that individual conformations can be isolated. These conformers are fundamentally different shapes, and thus often have different biological activities – in this case, it is atrop-abyssomicin C that packs the antibacterial punch.2
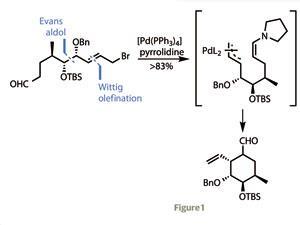
That potent biological profile, along with a structural Pandora’s box, has attracted a wealth of talented synthetic chemists. However, the routes developed thus far have limited potential in producing unnatural variants of the parent structure, hindering development of therapeutic analogues. Enter Filip Bihelovic and Radomir Saicic at the University of Belgrade, Serbia, with an extremely flexible route into this fascinating family.3
Their synthesis begins with a typical Evans auxiliary-mediated aldol coupling of nor-citronellal with a protected hydroxyacetaldehyde. After a bit of protecting group chemistry and a Wittig olefination to extend the carbon chain, one end was transformed into an allylic bromide, while the other was converted to an aldehyde. Treating this with a palladium catalyst and pyrrolidine prompted an organocatalytic Tsuji–Trost cyclisation, forming the cyclohexane ring in superb yield (figure 1).
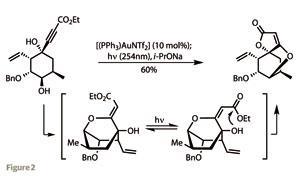
Excision of the aldehyde substituent followed, producing the corresponding cyclohexanone. This was alkylated with 3,3,3-triethoxyprop-1-yne under basic conditions to form a tertiary alcohol. After a little trial and error, the team arrived at a neat one-pot procedure to transform this monocyclic material into the complex tricyclic core of abyssomicin. Treating the acetylene with a gold(I) catalyst formed a complex with the triple bond. This could then be attacked by the hydroxyl group attached to the opposite carbon of the cyclohexane ring to close the second ring – a simple tetrahydropyran. However, as powerful as this reaction was, the olefin product had the wrong configuration to go on and form the desired spirocycle. Fortunately, irradiating the intermediate with UV light isomerised the double bond, and adding base prompted spirocyclisation (figure 2).
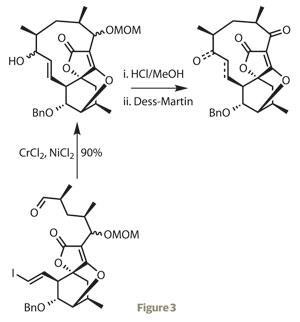
Having produced the intricate core of the target, the team moved on to constructing the surrounding macrocycle. They first alkylated the furanone ring with a sidechain constructed by a previously established route, then after a few steps for functional group interconversions, closed the 11-membered ring remarkably efficiently with a Nozaki–Hiyama–Kishi reaction (figure 3).
Oxidising both remaining hydroxyl groups left only one final deprotection step to deliver the target. However, closing down the macrocycle could theoretically happen with the sidechain passing over the top of the protruding carbonyl of the spirocyclic core, or underneath it. As the closed ring is too small to pass over this carbonyl, it is this that confers atropisomerism. The team found that only the desired atropisomer was produced by this reaction, and in good yield. Unfortunately, the paper does not discuss this outcome in any depth, but that should not detract from an impressive synthesis of an endlessly intriguing molecule.
Paul Docherty is a science writer and blogger based in Reading, UK
References
- B Bister et al, Angew. Chem., Int. Ed., 2004, 43, 2574 (DOI: 10.1002/anie.200353160)
- S Keller et al, Angew. Chem., Int. Ed., 2007, 46, 8284 (DOI: 10.1002/anie.200701836)
- F Bihelovic and R N Saicic, Angew. Chem., Int. Ed., 2012, (DOI: 10.1002/ange.201108223)












No comments yet