Although tricky to spot, rearrangements can simplify a synthesis, says BRSM
It’s rare these days for me to look at an organic molecule without quickly finding myself wondering how I’d make it, and starting to break it down in my head. I quickly learned during my training as a chemist that while reactions are usually taught in the forward sense, it’s at least as important to be able to think in the reverse direction and be able to plan a good synthesis. Unfortunately, this is a much harder skill to learn (or teach).
One research group I worked in would practice disconnecting molecules by dividing into small teams and holding monthly retrosynthesis competitions. The teams were given a target and asked to design the best route they could. They then presented this to the whole group, fending off criticism and defending their choices. It was fascinating to watch people propose completely different solutions to the same problem and – with no reactions off-limits – the breadth of chemistry you’d see in a meeting was vast. Despite this, there’s one large class of reactions that I don’t recall ever being discussed in these meetings – sigmatropic rearrangements. I would contend that this isn’t because they aren’t useful transformations, but because they’re not always immediately simplifying, and can be quite hard to ‘see’ during a retrosynthetic analysis.
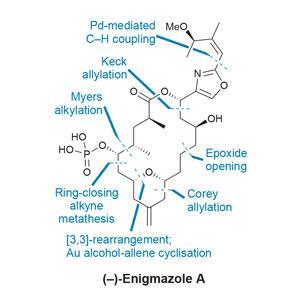
One good example of a simplifying late-stage sigmatropic rearrangement, which fits well into a complex route, comes in a synthesis of enigmazole A from Alois Fürstner’s group at the Max Planck Institute for Coal Research in Mülheim, Germany. The enigmazoles are a family of natural products that interfere with c-Kit signaling, an important pathway in cancer drug development that is also targeted by Novartis’ blockbuster leukaemia drug Glivec (imatinib). Several total syntheses of these bioactive natural products have already been completed.
The team assembles the precursor to the macrocycle using fairly well established chemistry, but towards the end of the route things start to get much more interesting. The macrolide ring itself is formed using a favourite reaction of the group – ring-closing alkyne metathesis. This allows direct access to the propargylic acetate needed for the planned rearrangement cascade.
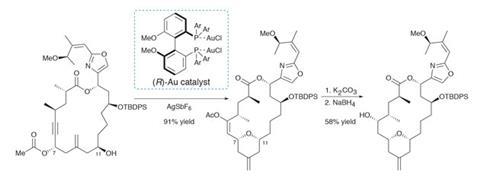
Treating this precursor with a chiral gold catalyst then kicks off a [3,3]-sigmatropic rearrangement (figure 1). Cyclisation of the nearby alcohol onto the resulting allene quickly follows (with a little help from the p-acidic gold). The reaction proceeds in excellent yield, and the resulting enol acetate is easily transformed into the final hydroxyl group. From here, phosphorylation and deprotection quickly complete the target. Using this transannular reaction to shrink the macrocycle is quite a clever tactic, as larger rings are generally less strained and hence easier to close – especially by metathesis.
A few follow-up studies on the mechanism of this key transformation reveal a number of surprising features (figure 2). Conventional wisdom holds that gold-catalysed rearrangements of propargylic acetates to allenes are both fast and reversible, and thus the stereochemical information in the starting alcohol is quickly lost, and should be inconsequential to the outcome.
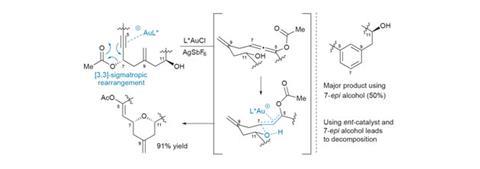
However, when the researchers start from the oppositely-configured (epimeric) acetate, they obtain only a trace of the desired product. Similarly, using the correct enantiomer of the gold catalyst is also crucial to maximise the yield of the reaction, with the opposite enantiomer giving inconsistent results and forming significant by-products. When the enantiomeric catalyst is paired with the epimeric starting material, no product is obtained at all, underscoring the importance of catalyst–substrate matching when applying asymmetric catalysis to chiral molecules. At this point it’s not clear if this strong stereochemical dependence is just a quirk of the molecule itself, but it’s an interesting discovery that, given the group’s interest in noble metal catalysis, they may well follow up.
BRSM is a process chemist and blogger based in the US
References
A Ahlers et al, Angew. Chem., Int. Ed., 2016, DOI: 10.1002/anie.201510026











No comments yet