Paul Docherty tackles a serious repeat offender
Those readers who have been subject to my rambling for more than a few months will note that my column tends to follow a general pattern – introduction to a molecule, its biological profile, structure and then the synthesis. This isn’t an accident – total synthesis papers in the literature tend to follow this same outline.
However, where trends exist, so do outliers. In a recent paper1 by Tohru Fukuyama and his team at the University of Tokyo, Japan, the target’s biological profile isn’t mentioned at all! This isn’t due to a lack of diligence on their part, or even on lack of biological activity on the part of the target – it’s that we’re dealing with a serious repeat offender.
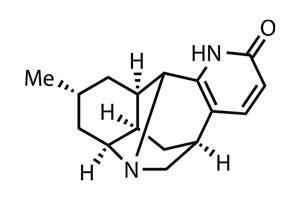
Since its isolation from the clubmoss Lycopodium complanatum in 2001, lyconadin?A has been party to three total syntheses (and has appeared in Totally Synthetic already as well). All that interest stems from anti-Alzheimer’s activity attributed to the lycopodium family of natural products and plants that give them their name.2
Introducing the fascinating structure of lyconadin A wasn’t particularly necessary either – the pentacyclic core, trisubstituted bridgehead nitrogen and 2-pyridone ring have all been discussed at length in previous papers, as has the synthetic challenge they present. What the Japanese team wanted to accomplish, though, was to build the complex cage structure as soon as possible to reduce the complexity that a late-stage cyclisation presents.
The synthesis begins with a very neat Diels–Alder reaction to add a unit of isoprene to a simple but enantiomerically enriched cyclohexenone.3 The reaction is directed by a methyl group, driving cycloaddition on the opposite face of the ring (figure 1).
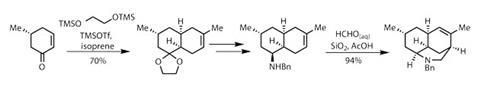
Cleaving the ketal and introducing a protected amine – by reductive amination with benzylamine – sets up an ideal substrate for an aza-Prins cyclisation. Treating with formalin (aqueous formaldehyde) under mildly acidic conditions allows the reaction to proceed, joining the amine to the alkene to make a third ring.
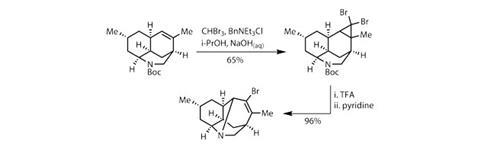
After a quick break to swap protecting groups on the tertiary amine, the team was ready to complete the complex pentacyclic cage system. The plan was to use a ring expansion, in which a cyclopropane ring would be induced to rearrange, forming a carbocation that could be quenched by the nitrogen.
Using bromomethane, the team constructed the strained three-membered ring on the cyclohexene, then removed the butoxycarbonyl protecting group using trifluoroacetic acid. Heating the strained system with pyridine prompted the expected electrocyclic ring opening and rearrangement, expanding one ring and closing another to form the cage structure in remarkable yield (figure 2).
One of the benefits of this strategy is that the cyclisation leaves a useful functional handle – a vinyl bromide. After investigating several options, the team evolved a high-yielding method to transform this into an even more useful enone. Converting the vinyl bromide into the corresponding vinyl azide is relatively straightforward, followed by gentle heating in aqueous acid to release nitrogen and then hydrolyse the resulting imine to leave the enone.
The team’s desire for an enone was understandable, as their final task was to assemble a pyridone. Routes towards pyridones are diverse, but enones are uniformly suitable starting materials. An amide side chain was added using 2-(phenylsulfinyl)acetamide: deprotonating between the electron-withdrawing carbonyl and sulfinyl groups produced an ylide, which undergoes conjugate addition into the enone (figure?3).
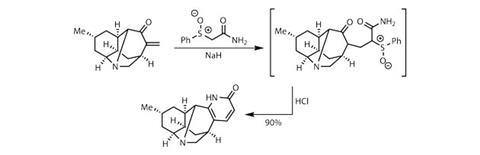
Adding a little acid to the same flask then prompted cyclisation, completing the target in a rather efficient 12 steps.
Paul Docherty is a science writer and blogger based in Reading, UK












No comments yet