Exceptions to a long-held rule against chemically bonding to biological targets are powering new cancer medicines, finds Andy Extance
From its discovery in the late 1800s until the 1970s, the humble aspirin kept a secret hidden that would have unnerved medicinal chemists. Not only does its active component, acetylsalicylic acid, dock with cyclooxygenase enzymes COX-1 and COX-2 and stop them making the prostaglandin molecules behind inflammations – it bonds to them covalently and permanently. Such irreversibility has long made drug developers anxious, in part due to another medicine cabinet stalwart, paracetamol. Our bodies metabolise paracetamol, also known as acetaminophen, to make a highly reactive intermediate that covalently binds to liver proteins. This has been linked with toxicity, and fear of similar problems has driven an orthodoxy in drug discovery to avoid drugs that attach permanently.
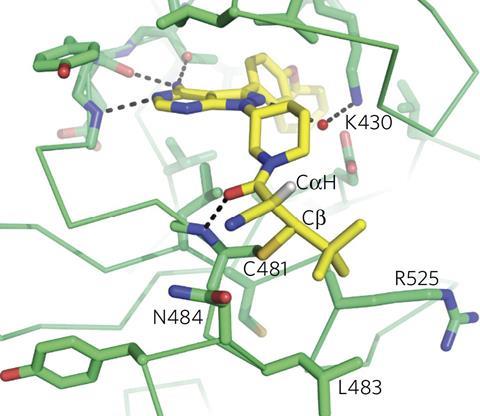
Today, that orthodoxy lies shattered. Several recent studies have shown that there are many covalent drugs, as they are known, despite the bias against them. For example, one study analysing 71 enzymes found around a quarter were covalently inhibited by approved drugs.1 In 2011, researchers estimated that 26 covalent drugs for which data were available accounted for over $33 billion (£26.4 billion) in annual worldwide sales.2 Consequently, drug developers have come to realise that covalent binding is more than just allowable – in some circumstances it’s actually desirable. ‘We’re only now trying to design them rationally, rather than be limited to discovering them serendipitously,’ admits Renato Bauer, a medicinal chemist at Eli Lilly in Indianapolis, US.
Over the past five years those efforts have already paid dividends, with five intentionally covalently bonding cancer drugs hitting the market. Yet much remains for scientists like Bauer to do, including bringing the approach to diseases that can’t tolerate off-target effects like those that originally caused alarm.
Jamming the lock
If enzymes are locks and the molecules that bind with them are keys, covalent inhibitors effectively snap the key off in the lock. The permanent obstruction is an advantage, underscores David Goldstein, chief scientific officer at Principia Biopharma in San Francisco, US. Such molecules can turn off enzymes in one hit, at least until our cells resynthesize them, which can happen over a timescale of several hours to a few days. Therefore patients should need to take less of the drugs.
Enzyme-lifetime permanence, and the advantage covalent attachment has in competing with enzymes’ normal partners, also means drug molecules might be designed so any excess leaves the body quicker. And while non-covalent drugs rely on binding site features that are usually similar in different enzyme family members, covalent attachment happens at reactive sites that are not always shared. Consequently, covalent inhibitor drugs could be more selective. Overall, these advantages could minimise patients’ exposure to drugs, any side-effects they might cause, and also interactions that could lead to resistance.
‘Principia was founded to expand upon that concept to create small molecule drugs that could achieve greater selectivity and longer target engagement than traditional non-covalent methodologies,’ Goldstein explains. ‘Such an approach was envisioned to realise drugs with both greater efficacy and tolerability.’ And appropriately, the primary drug class scientists using this expectation-upending approach have focused on – kinase inhibitor cancer drugs – has itself upended expectations.
Not undruggable, but unpleasant
Kinases are enzymes vital to many biological processes, involved in processing ATP, the ‘energy currency’ molecule. Because humans have a ‘kinome’ of over 500 kinases with similar active sites in which drugs would bind, they were once seen as ‘undruggable’. If a drug blocks one kinase type to stop cancer cell metabolism then it’s likely to block others, with potentially serious adverse effects. Nevertheless, in 2001 tyrosine kinase inhibitor (TKI) imatinib (Glivec), was approved for chronic myelogenous leukaemia, a rare cancer. And today, for the improved survival chances various TKIs now on the market bring, cancer patients are prepared to accept their unpleasant downsides, which can include severe diarrhoea.
‘Selective targeting of a small number of kinases remains one of the major challenges,’ Goldstein says. ‘This problem is particularly acute within the stomach and upper gastrointestinal (GI) tract where drug concentrations are often very high following an oral dose. At those concentrations, poorly selective TKIs can inhibit a significant fraction of the kinome expressed in the GI tract, leading to severe and often dose-limiting toxicities.’
By contrast, targeting cysteine amino acid residues through covalent binding enables TKIs to achieve ‘unprecedented selectivity and potency’, Goldstein asserts. Principia is using this approach in PRN1371, a TKI targeting the fibroblast growth factor receptor currently in phase I trials. Covalent drugs have also been used to inhibit epidermal growth factor receptor (EGFR) tyrosine kinases when they have mutated and made cancers resistant to non-covalent drugs.
It requires great precision to shape molecules so that they attach only to reactive sites on one enzyme type and avoid others. To achieve this, Pfizer has taken a detailed approach to using and studying covalent inhibitors, explains Adam Gilbert, the senior director of design and synthesis sciences at the company’s Groton site in the US. ‘We decided to pursue covalent inhibitors for targets/programmes where they would offer a distinct advantage over reversible inhibitors,’ Gilbert explains. ‘We also decided that understanding the reactivity of the covalent inhibitor electrophiles with biologically relevant nucleophiles would give us insight into covalent inhibitor design.’
Pfizer’s studies include analysing the intrinsic reactivity of covalent inhibitor electrophiles with glutathione3 and N-acetyl-lysine4, to model cysteine and lysine amino acid nucleophiles they might target respectively. ‘We subsequently used this data to inform the design of covalent inhibitors for a number of past and current programmes by using the least reactive electrophile to give us the potency and selectivity we desired,’ Gilbert says.
With improving covalent kinase inhibitor selectivity the greatest challenge Pfizer initially faced, Gilbert adds, it joined forces with Ben Cravatt at the Scripps Research Institute in San Diego, US. Cravatt had been drawn to covalent inhibitors as a more rigorous tool for understanding how small molecules interact in biological systems. ‘Because they remain permanently attached, you can inventory proteins using mass spectroscopy-based methods,’ he explains. Among the details listed in such inventories are the exact locations where covalent attachments are happening.
Testing warheads
For example, Cravatt’s team and Pfizer teams cultured cancer cells with covalent TKIs. Smashing the cells open and analysing fragments of the proteins inside identified 29 off-target proteins that inhibitors attached to.5 The study revealed specific TKI ‘concentration windows’ for selective target inhibition beyond which they reacted with off-target proteins, with toxic effects.
The results also showed covalent attachments are frequently not actually in the binding pocket ‘keyhole’ enzyme inhibitors normally target, Cravatt says. ‘Every drug that I’m aware of today that’s covalently targeting cysteine is actually targeting non-catalytic, bystander cysteines on the outside of the protein,’ he says. ‘They’re reasonably selective, but most of their off-targets aren’t kinases, they’re other proteins that harbour functional cysteine residues.’
At Scripps’ site in Florida, US, scientists are exploring whether in some instances covalent inhibition might work in reverse. Kate Carroll’s group found that some cancer cells became resistant to Boehringer Ingelheim’s covalent TKI afatinib (Giotrif) due to the oxidation of its target cysteine in EGFR. This happens because there are lots of reactive oxygen species in cancer cells, which can convert cysteine to its sulfenic acid (Cys-SOH), explains team member Vinayak Gupta.
Rather than a nucleophile like cysteine, the sulfenic acid is an electrophile, which may help explain other compounds’ biological activity. ‘The idea here is that there are “nucleophilic” compounds already “hidden” in current natural products such as curcumin and FDA-approved drugs such as [Pfizer’s TKI] tofacitinib,’ Gupta says.6 ‘It is a distinct possibility that there may be an underlying mechanism of action driven by these “hidden” nucleophiles targeting Cys-SOH in their biological targets.’
Consequently, Carroll, Gupta and their colleagues have sought to profile the molecules that could exploit this phenomenon. But initially, in contrast to the relatively broad range of electrophile inhibitors available, there was only one moderately reactive nucleophile available to detect Cys-SOH. The Scripps Florida team have therefore developed a library of potential covalent inhibitors. ‘We now have nucleophilic warheads that react with Cys-SOH at biologically relevant rates, more than 200 times faster than the previous standard for Cys-SOH probes, both irreversibly and reversibly,’ Gupta says.7 That’s especially important as scientists seek to understand how to sort such arsenals, eliminating cruder blunderbusses and getting closer to the proverbial magic bullet.
Toxicity dumped?
Despite their advantages, covalent drugs have seldom been developed for diseases other than cancer, except in older drugs where the covalent effect was introduced accidentally. Outside of life-or-death diseases, the risk of adverse effects caused by irreversible modification from off-target nucleophiles has been deemed too great.
Highly reactive, poorly selective covalent binders, for example, ‘could certainly cause toxicity issues’ by raising protein molecular weights above the threshold where they trigger immune responses, says Gilbert. He and his Pfizer colleagues therefore treated human liver cells with radiolabelled covalent drugs in the lab to estimate the ‘covalent binding burden’ limit that might be allowable.8 ‘We found examples of safe covalent drugs which have been used for decades with a covalent binding burden of less than 10 mg/day,’ Gilbert says. ‘While one can’t say if 10 mg/day is the magic binding burden limit for a safe agent, we believe the binding burden assay can be used to optimize covalent binders towards safer space.’
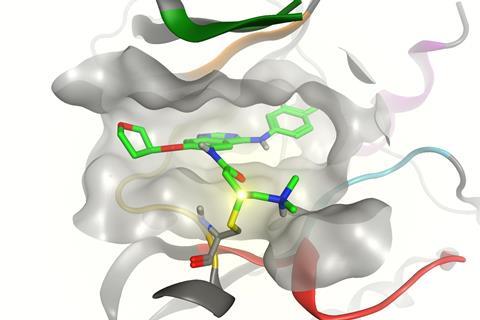
By working to ensure their drugs are selective enough, various companies are now able to put low dose covalent inhibitors into clinical trials. ‘These compounds are showing good safety profiles,’ Gilbert says. ‘One can’t rule out or predict idiosyncratic toxicity with these agents, but so far this well-designed new generation of covalent inhibitors shows similar toxicity profiles to reversible inhibitors.’
That might be acceptable when fighting cancer, but it still means covalent inhibitors are challenging to develop for chronic non-life-threatening diseases, Goldstein asserts. Consequently, Principia Biopharma is exploiting a concept pioneered by its co-founder, Jack Taunton of the University of California, San Francisco (UCSF). His team discovered that electrophiles like cyanoacrylamides could bond to cysteine residues covalently – but unlike other existing drugs, they do so reversibly.9
The consequence of any off-target binding with compounds like cyanoacrylamides should be transient, Goldstein explains. ‘With reversible covalent inhibition, we have demonstrated that proteins regain their function when the inhibitor dissociates from its target, not permanently altering their structure and function,’ he says. ‘As such, the consequence of reversible covalent binding is not different from non-covalent small molecule drugs.’
Principia uses the approach to target Bruton’s tyrosine kinase (BTK), a key element in human immune signalling pathways, involved in diseases like rheumatoid arthritis.10 Currently the company is running phase II trials in the rare and potentially life-threatening skin-blistering disease pemphigus vulgaris. ‘In autoimmune disease, Principia’s lead molecule, PRN1008 is a reversible covalent inhibitor that inhibits 100% of BTK initially after oral dosing,’ Goldstein explains. ‘It slowly comes off the target resulting in approximately 60-80% inhibition prior to the next dose despite high clearance of the compound. This profile achieves high efficacy combined with good tolerability and safety.’
Bound for success?
For medicinal chemists at Eli Lilly, Bauer says, interest in covalent inhibitors extends broadly – not only to electrophiles that react reversibly, but also to electrophiles for which little is currently known in drug discovery. Lilly also has a irreversible covalent BTK inhibitor in phase II trials for rheumatoid arthritis. It appears that conventional design of properties such as molecular shape can produce sufficient selectivity to avoid off-target binding in cases like this. ‘Whether moderate electrophiles, such as acrylamides, are disposed to indiscriminate alkylation seems to depend more on the properties of the parent molecule rather than the simple fact that an electrophile is present,’ Bauer explains.
Like any individual drug, covalent inhibitors still face high chances of failing to secure regulatory approval. Nevertheless, this new approach could have an even broader and fundamentally important impact. ‘Covalent binding is also proving important in other areas of discovery, like in chemical biology approaches to understanding disease,’ Bauer explains. ‘For early stage projects, covalent inhibitors play additional roles, such as tool molecules to elucidate the effect of perturbing a biological pathway. Covalent inhibition is unique and complementary to many conventional approaches.’
Perhaps covalent inhibitors’ greatest opportunity is exemplified by another non-kinase target that Bauer highlights. K-Ras(G12C) is a mutated GTPase protein, involved in cell signalling, which occurs in about 20% of lung cancers. However, it ‘has historically been viewed as something that’s very difficult to drug’, cautions Cravatt. Yet by exploiting reactive cysteines, in 2013 Kevan Shokat’s UCSF team found compounds that bind it.11 These covalent inhibitors exploit their ability to attach outside the conventional binding pocket ‘keyhole’, in a previously unknown site.
And Cravatt’s team has found scope for emulating this sneaky lock-picking approach elsewhere. They have screened small electrophiles in cancer cells with the help of an extremely reactive iodoacetamide electrophile, which reacts with every available cysteine, unless blocked by another compound.12 The Scripps researchers then added isotopic tags to the iodoacetamide residues, which allowed them to quantify their relative abundance. ‘You’re assessing 50–100 enzymes, in a mechanistically related class,’ Cravatt explains.
Their results indicate that – as in UCSF’s K-Ras(G12C) work – covalent inhibitors often target ‘totally different folds, totally different functions’, Cravatt explains. He’s now exploring this phenomenon more broadly, including through Vividion Therapeutics, which he co-founded with fellow Scripps chemists Jin-Quan Yu and Phil Baran in February 2017. A substantial component of Vividion’s activity will be covalent chemistry-driven, Cravatt reveals.
Cravatt is especially excited by covalent inhibitors’ prospects because ‘there have been multiple successes in recent years’. ‘Covalent chemistries developed for kinase inhibitors, you can make a strong argument that they have an advantage over reversible kinase inhibitors,’ he says. Yet he’s keen to go beyond kinases’ ‘very druggable pockets’. ‘The question is can we progress against proteins viewed as undruggable with non-covalent chemistry?’
Andy Extance is a science writer based in Exeter, UK
References
1 J G Robertson, Biochemistry, 2005, 44, 5561 (DOI: 10.1021/bi050247e)
2 J Singh et al, Nat. Rev. Drug Discov., 2011, 11, 307 (DOI: 10.1038/nrd3410)
3 M E Flanagan et al, J. Med. Chem., 2014, 57, 10072 (DOI: 10.1021/jm501412a)
4 U P Dahal et al, Med. Chem. Commun., 2016, 7, 864 (DOI: 10.1039/c6md00017g)
5 B R Lanning et al, Nat. Chem. Biol., 2014, 10, 760 (DOI: 10.1038/nchembio.1582)
6 V Gupta and K S Carroll, Chem. Commun., 2016, 52, 3414 (DOI: 10.1039/c6cc00228e)
7 V Gupta and K S Carroll, Chem. Sci., 2016, 7, 400 (DOI: 10.1039/c5sc02569a)
8 U P Dahal et al, Chem. Res. Toxicol., 2013, 26, 1739 (DOI: 10.1021/tx400301q)
9 I M Serafimova et al, Nat. Chem. Biol., 2012, 8, 471 (DOI: 10.1038/NChemBio.925)
10 J M Bradshaw et al, Nat. Chem. Biol., 2015, 11, 525 (DOI: 10.1038/nchembio.1817)
11 J M Ostrem et al, Nature, 2013, 503, 548 (DOI: 10.1038/nature12796)
12 K M Backus et al, Nature, 2016, 534, 570 (DOI: 10.1038/nature18002)










No comments yet