The 2012 Nobel prize in chemistry was awarded to Robert Lefkowitz and Brian Kobilka for studies of G-protein-coupled receptors. Phillip Broadwith looks at the molecular machinery underpinning cell signalling
The story of some Nobel prizes starts with a single eureka moment, an epiphany that shifts the course of a particular researcher’s enquiries in new directions. Not so for this year’s prize in chemistry.
The struggle to understand and visualise the complexity of G-protein-coupled receptors (GPCRs) and their effects on almost every cell of our bodies has taken decades. The steps along the way have, at times, been agonisingly small; each fragment of progress has had to be pieced together until the picture finally began to emerge.
It has taken extraordinary passion, commitment and dogged determination on the part of both this year’s laureates, Robert Lefkowitz of the Howard Hughes Medical Institute and Duke University in Durham, US, and Brian Kobilka of Stanford University, US, as well as their many students and collaborators.
But their work has opened a door to understanding the way information outside of cells – in the form of hormones, neurotransmitters, drug molecules and the myriad other signalling molecules in our bodies – translates into action inside cells. The GPCR protein family is vast, with around 800 known in humans, around half of which are the olfactory receptors that give us our sense of smell. The other half respond to signalling molecules like adrenaline, dopamine and histamine, as well as certain lipids and peptide hormones. These molecules stay outside the cell, but changes to the conformation of the GPCR trigger different internal responses mediated by a range of G-proteins.
‘What they’ve done is give us a complete molecular understanding of one of the most important classes of proteins,’ says Fiona Marshall, chief scientific officer of GPCR-based drug discovery company Heptares. ‘Pretty much every disease you can think of, there’s a GPCR that could be a target for that disease. Around 40% of drugs currently on the market work through this family of receptors,’ she adds.
Fathers of the field
‘When you come into a field like this, there are some people who just seem to have been in the field forever, like Lefkowitz – he seems to have done everything!’ says Chris Tate, who researches GPCRs and other membrane proteins at the Medical Research Council Laboratory of Molecular Biology (LMB) in Cambridge, UK. ‘He was involved in cloning the receptors, as well as sorting out the pharmacology and the biochemistry. And Kobilka was involved right from the beginning too, with the cloning of the first receptors – so both of them are real fathers of the field.’

The magnitude of the problem becomes clear as Lefkowitz explains the steps that were involved: developing radiolabelled ligands to bind the receptors, as well as tools to tag and track their activity; followed by purifying the first receptors. ‘That was unbelievably difficult and took a decade,’ he says. ‘It required not just a way to solubilise the receptors, but anywhere from a 200,000- to 400,000-fold purification – these proteins are truly trace contaminants of plasma membranes. We never had more than 25 or 50µg of pure receptor at any time, because it was so difficult to make.’
But eventually they did get enough pure receptor to be able to fragment them and work out the protein sequence. From that, they could get to DNA sequences and the genes that encoded the receptors so that they could be cloned and expressed in large enough quantities to be crystallised. ‘Each of these steps was confounded by all manner of technical hurdles,’ says Lefkowitz. ‘It was a bear at every stage, and that’s continued into the present.’
But once that initial isolation work was done, the close relationships between the whole family of GPCRs could be exploited to search for similar sequences. ‘To start with, there was no way in [except by purifying the native receptors],’ Lefkowitz explains. ‘But by getting those pieces of sequence, we could clone the first few receptors, and then those were like the Rosetta stone – people could use homology techniques to, little by little, expand the family.’
Crystal conundrums
While Lefkowitz and his team concentrated on cracking the biochemistry and pharmacology of how the GPCRs function, one of the post-doctoral fellows involved in that cloning effort, Brian Kobilka, became fixated on getting a glimpse of their structures.
‘Once he left Bob’s lab, it was clear he was on a crusade to get the 3D structures of these receptors,’ says Michel Bouvier, who overlapped with Kobilka as a post-doc with Lefkowitz, and now studies GPCRs at the University of Montréal in Canada. ‘Almost everything they do in his lab has something to do with getting that structural information.’

Bouvier explains that one of Kobilka’s greatest strengths is in forging collaborations, tapping in to some of the foremost experts in various fields, then applying that expertise to the GPCR problem – how to modify the proteins to make them more stable; how to purify them to get large quantities; how to crystallise them and then working with the best crystallographers to get good structures. ‘But he was the force behind the whole thing,’ adds Bouvier
He also points out that Kobilka’s single-minded pursuit, which took around 15 years before it actually yielded a successful crystal structure, could potentially have been his downfall. ‘It was a big risk that he took; at times he lost a lot of his funding, the lab could have been in big trouble,’ says Bouvier. ‘He took big gambles, and the fact that he has pulled through like this has made a lot of people very happy – Brian is a person that everybody likes.’
Kobilka himself is quick to emphasise the role that other people have had in keeping him going. ‘Having a supportive wife, who has also been a partner in science, is probably the most important factor in moving forward when things weren’t going well,’ he says. ‘I have also been very fortunate to have great colleagues, students and postdoctoral fellows.’ Kobilka’s wife, Tong Sun, who trained as a medical doctor, works alongside him at Stanford, and is chief executive of the spin-out company that they co-founded, ConfometRx.
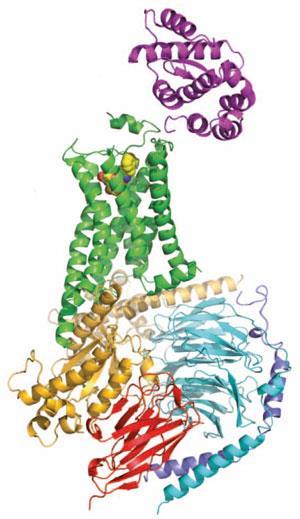
Some of the strategies that Kobilka’s group have used to study the structural biology of GPCRs have turned out to be remarkably general. Tate points out that one of the loops between the characteristic helices that form the membrane-spanning portion of the receptors can be particularly troublesome in crystallisation, because it is often rather flexible. Replacing that section of the protein with the sequence from an enzyme called T4 lysozyme has proved to be a tremendously successful strategy and led to several receptor structures.
But while Kobilka has led the way in crystallising GPCRs since the publication of the first structures in 2007, it was his team’s publication of an adrenaline receptor complexed to its G-protein signalling partner that stood out. ‘That is the thing that really sets Brian head and shoulders above everybody else in the field,’ says Tate. ‘I really don’t know how many times it will be repeated, because it’s really tough to do.’
Tate explains that the G-proteins themselves, when they bind to the receptor, are incredibly unstable, so the team had to use a fragment of an antibody from a llama to stabilise the interface between the three subunits of the G-protein to get it to crystallise. ‘It really shows for the first time how an activated GPCR can activate a G-protein,’ he says.
Only half the story
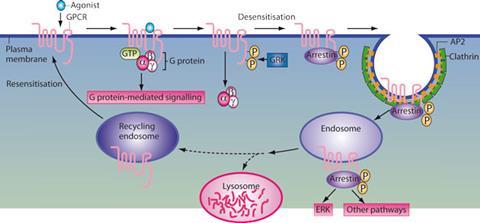
If G-protein signalling were the only pathway activated by GPCRs, that would still be remarkable. However, focusing solely on the G-proteins is to miss an important facet of the receptors’ biochemistry. ‘When activated, a GPCR interacts with the G-protein. It also becomes phosphorylated on multiple sites, which leads to binding of a beta-arrestin and shuts the G-protein signalling off,’ explains Lefkowitz. This is also the mechanism by which receptors become desensitised to stimuli – even if the concentration of the signalling molecule outside the cell stays high, within a few minutes the G-protein activity will stop.
But Lefkowitz and his team discovered that the beta-arrestins can also trigger signalling pathways of their own. Not only do they shut off G-protein signalling, but they can activate signalling cascades that are completely independent of G-proteins, or involve a combination of both G-proteins and arrestins.
Lefkowitz also began to find molecules that could selectively turn on one or other of these pathways – so-called ‘biased’ ligands. Realising the possible pharmacological implications, he set up a spin-out company called Trevena with a couple of his post-docs to explore it further.
Into industry
Jon Violin, one of those founding post-docs and now Trevena’s head of biology, explains that the company uses proprietary assays to screen for compounds that selectively turn on one pathway or the other. They follow this with a classical iterative medicinal chemistry approach to improve their selectivity and potency, and also develop structure–activity relationships to guide further studies.
Violin explains that in some diseases, the desired medical effect is associated with one pathway, and the side effects are mostly triggered by the other, so a biased ligand would be a much better drug. For example, Trevena has a compound called TRV130, now in Phase I clinical trials, which targets the mu-opioid receptor and selectively turns on only the G-protein pathway. ‘Opioid analgesics like morphine have a host of adverse effects,’ says Violin. ‘They cause respiratory depression (that’s what leads to death in cases of overdose), gastrointestinal dysfunction – nausea, vomiting, constipation – and obviously they are addictive as well.’
Tests on mice lacking the beta-arrestin pathway indicated that most of those side effects are mediated by beta-arrestin signalling, Violin says. ‘So if you could find a drug that mimics morphine in terms of its analgesic effect, which we know is mediated by G-protein coupling, but avoids beta-arrestin, then you might have a better drug. That’s what we’ve found in TRV130 – in pre-clinical studies it’s a very powerful analgesic, with reduced respiratory depression and constipation.’
But Violin also points out that sometimes it’s completely the other way around. Another Trevena compound, TRV027, selectively turns on beta-arrestin signalling at the angiotensin receptor. The inspiration for this programme came from angiotensin receptor blockers (ARBs) which are widely used to control high blood pressure (hypertension). ‘When someone is given an ARB, we know that that has clinical benefit by blocking angiotensin from binding the receptor, [which stops G-protein activity],’ he explains. ‘But ARBs also prevent angiotensin from turning on beta-arrestin pathways, and some of those are good.’
We’re using those structures in virtual screening, drug design and lead optimisation
Structural data is also guiding efforts to develop new and better drugs to target a range of GPCRs. ‘If you’re in the pharmaceutical business, you want to know the structure of every GPCR you’re interested in, and preferably multiple structures with different ligands bound, because you’re interested in how you can change those ligands to affect efficacy or selectivity,’ says Tate.
This is what UK company Heptares, which was founded by a group of scientists from the LMB in Cambridge, including Tate, is trying to do. ‘We’ve developed our own way of getting x-ray structures of GPCRs, slightly different from the approach that Brian Kobilka has been taking,’ says Heptares’ Marshall. ‘It involves engineering the proteins to make them more stable, so they’re not dependent on the membrane environment.’ This means the proteins can be isolated, purified and used in fragment screening for drug discovery, or crystallised to determine their structures. ‘We’ve got a large number of structures now, and we’re using those structures in virtual screening, drug design and lead optimisation,’ says Marshall. ‘We’ve been able to use the knowledge of the structures to actually get selectivity where that hasn’t been possible before.’
Tate believes that the transition of GPCR structural biology from a purely academic to an industrial setting marks a turning point. ‘Industry is usually fairly conservative about challenging projects like this, but it’s now being done, and that’s really exciting,’ he says.
Together again

Both Lefkowitz and Kobilka are also enthusiastic about the future; in fact, the two groups have recently begun collaborating again. Part of that collaboration is an attempt to crystallise the angiotensin receptor, and as Lefkowitz explains, this highlights the fact that while many of the techniques Kobilka has introduced are reasonably general, there is still no guarantee of a successful structure. ‘We’ve been working together on it for 3–4 years, and it just won’t work. We’ve tried every trick in the book,’ he says. ‘It would be gorgeous, because we have totally G-biased and totally beta-arrestin-biased ligands for it, but we just can’t crystallise the darn thing!’
Another goal is to solve structures of GPCRs coupled to beta-arrestins. ‘We still have a lot to learn about how specific GPCRs couple to specific G-protein subtypes, as well as to other signalling and regulatory proteins,’ says Kobilka. ‘Some of these questions may be answered by crystallography; however, it may not be possible to crystallise all of the functionally relevant interactions, so we will need to apply other biochemical and biophysical approaches.’
The struggle to decipher the mysteries of G-proteins is far from over, and there will no doubt be significant challenges on the way. And for Kobilka, that is one of the biggest motivators. ‘I believe most scientists enjoy the process of tackling a difficult problem almost as much as achieving the goal,’ he says, ‘and we often learn something from our failures that help us move forward.’
A family affair

Speaking to Lefkowitz, it is clear that this year’s Nobel prize is recognising not only great science, but also great mentorship. A significant number of the world’s top GPCR researchers have trained with Lefkowitz, Kobilka or both. Lefkowitz explains that he has become more and more conscious of the importance of mentorship over the course of his career. ‘When it comes to mentoring, like everything else, you have to play to your own strengths. I know that Brian is viewed as a wonderful mentor to his students and fellows, but his personality and mine couldn’t be further apart, so there’s no way we can mentor in the same way. But that doesn’t mean we can’t both be very good mentors.
‘Part of my personality is that, to me, my lab is like a second family,’ Lefkowitz adds. ‘I enjoy trying to figure people out, and figure out what I think their potential is. Then I take it almost as a personal challenge to see if I can get each and every person, at least sometime while they’re with me, to see what it’s like working right at their best level of activity. You have to work differently with different people depending on what their natural gifts are.
‘The fact that I’m sharing this prize with Brian makes it so much better than if I was getting it alone, or sharing it with someone else who didn’t train with me. It feels perfect,’ he says.












No comments yet