The chemistry of making – and losing – memories is increasingly well understood, as Rachel Brazil discovers
Manipulating memory is a popular theme in science fiction and while the plots of films like Eternal sunshine of the spotless mind and Total recall might be far-fetched, recent advances are making memory enhancement more than just fantasy. Since the early 1990s, neuroscientists have been piecing together the complex jigsaw of how we establish and lose memories, and this growing molecular understanding could soon be used to help those suffering from neurodegenerative diseases or battling drug addiction.
The first genetically modified mice with improved learning and memory were created in 1999 by Joe Tsien, a neurobiologist at Princeton University in the US. He named the new strain Doogie, after the precocious teenage TV doctor. The animals could solve mazes in half the usual time and retained this knowledge for longer than usual. The Doogie mice had an extra copy of the NR2B gene – responsible for producing the protein NMDA (N-methyl-d-aspartate), a receptor for the signalling chemical glutamate.
The Doogie mice confirmed the importance of NMDA receptors for learning and memory via a mechanism almost identical in humans and rodents – long term potentiation (LTP). It leads to stronger responses with repeated stimulation of neural connections by remodelling and enlarging synapses – the small gaps between neurons. Neurons have tendrils called dendrites at one end where information is received, and a long fibre, the axon, whose tip releases chemicals called neurotransmitters. These cross the synaptic gap and bind to receptors in neighbouring cell membranes, setting off further signalling cascades.
Simply put, learning involves the association of two events. The importance of NMDA is that it provides a way of doing this by operating only when it receives signals from two neurons. At the centre of the receptor there is a channel, blocked by a magnesium ion. External stimulation causes depolarisation, where sodium ions enter the neuron and instantly increase the voltage. The process cascades along the cell to the synaptic terminal, releasing glutamate molecules that cross the synaptic gap to NMDA receptors. When stimulated by two neurons, the magnesium ion shifts, flooding the cell with calcium ions. This triggers a cascade of intracellular signalling, ultimately leading to growth of the synapse and additional NMDA receptors. These enlarged synapses are the physical basis of new memories.
Controlling memory with epigenetics
One unanswered question is how the brain maintains a memory for long periods of time, when the proteins involved in their storage deteriorate. ‘You need some sort of long lasting or perpetuating mechanism for maintaining memory traces if the components of that memory are being degraded,’ says Courtney Miller, neurobiologist at the Scripps Research Institute in Florida, US. Miller worked on this question as a researcher in the lab of David Sweatt at the University of Alabama at Birmingham, US, and it led them to consider the role of epigenetics. Epigenetics explains how cells switch genes ‘on’ and ‘off’ to produce different proteins. One mechanism is DNA methylation. When some cytosine bases are methylated at the five position of their aromatic ring, access to the DNA is blocked and a gene can be turned off. The absence of methyl groups has the opposite effect, switching a gene on.
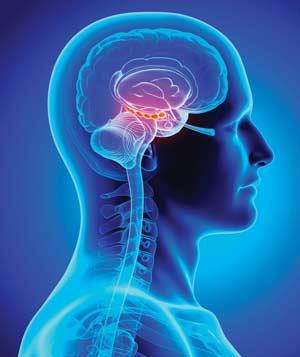
In 2007, Miller and Sweatt showed that the enzymes responsible for DNA methylation, DNA methyltransferases (DNMTs), were increased in the hippocampus of a rat’s brain following learning, and pinpointed a number of genes also rapidly turned on and off by DNA methylation.1 The hippocampus is a region deep inside the brain identified as important for memory. Miller and Sweatt tested rats’ learning using fear-conditioning experiments, where the animals were trained to associate a particular environment with electric shocks. Subsequently, if a fear memory formed, the rats froze when introduced into that environment. They found that memory formation could be stopped by giving the rats chemicals that block the action of DNMTs. These DNA methylation changes lasted just a day, suggesting that only initial memory storage occurs in the hippocampus. Further experiments one month after learning showed increased DNA methylation across the cortex – the outermost layer of the brain, where longer-term memories are thought to reside.2
Another epigenetic mechanism for controlling gene expression is histone acetylation and in 2007, Li-Huei Tsai and Andre Fischer from the Massachusetts Institute of Technology in the US showed it also has a role in learning and memory.3 Histones are the proteins around which DNA is wrapped in cell nuclei forming a complex known as chromatin. The addition or removal of acetyl or methyl groups to lysine amino acids on histones controls the accessibility of DNA for copying, thus turning genes on or off.
Tsai and Fischer were looking for ways to improve learning and memory for sufferers of neurodegenerative diseases. They first showed that an increased learning ability is linked to histone acetylation in the hippocampus. Then, by inhibiting histone deacetylases (HDAC), the enzymes that remove acetyl groups from histones, they were able to improve the damaged cognitive abilities of mice genetically engineered to model the progression of Alzheimer’s disease.
In fact, as well as slowing memory deterioration, HDAC inhibitors seemed to allow animals to retrieve lost memories. Tsai and Fischer took Alzheimer’s model animals that had gone through fear conditioning prior to the onset of memory loss and gave them an HDAC inhibitor for one month. They found their abilities to recall the fear conditioning returned. Exactly how this happens is not clear, but Tsai has a theory. ‘During neurodegeneration, connectivity is impaired so memories cannot be retrieved. HDAC inhibitors ` increase synaptic density, thus may repair the brain circuits and facilitate remote memory recall.’
Miller also investigated the role of another epigenetic control system, microRNA – small strands of RNA, around 22 basepairs long. These molecules block protein production giving another way of regulating genes. ‘MicroRNAs are appealing because of their ability to potentially regulate hundreds of different gene targets – you might be able to have a really profound effect on memory,’ she explains. Miller found changes in microRNA levels in rodents’ amygdalas, the brain’s emotional memory centre. In particular, after learning, microRNA-182 decreased to nearly undetectable levels. The molecule was identified as a repressor of the protein actin, which is part of a cells cytoskeleton, but also has an important role in the remodelling and growth of the synapses. The globular monomer form, G-actin, forms polymer microfilaments called F-actin that can branch and form spine-like structures causing enlargement of the synapse. The absence of microRNA-182 promotes actin synthesis and strengthens the synapse connections necessary for memory formation.4
The role of epigenetics may also explain why memory deteriorates with age. A growing body of research is showing that epigenetic controls are lost with age, leading to cancer and diabetes, for example. So can memory loss be attributed to a similar process? According to Tsai, when mice age, the expression of genes responsible for learning and memory is drastically reduced, as is histone acetylation around those genes. ‘As you age, if microRNA-182 levels go up, it might be a lot harder to suppress them sufficiently to allow memory formation,’ Miller suggests, but she adds this is just speculation at the moment.
Active forgetting
Neuroscientist Attila Stetak from the University of Basel in Switzerland has been studying the other side of the coin – forgetting. Stetak says its not just a passive process where memories are overwritten or decay but that biochemical controls are involved, which he calls ‘active forgetting’. Stetak’s discovery, published in March this year,5 came from experiments with roundworms. Their memory can be tested by conditioning them to associate a chemical such as diacetyl with starvation – the memory will lead them to avoid diacetyl in future experiments for a period of time. Stetak found that worms genetically engineered without a protein called musashi seemed less forgetful and kept the associative memories longer.
Stetak suggests that glutamate receptors initiate mechanisms for memory formation and forgetting. ‘It’s like a switch that turns on both sides, so storage and removal of storage.’ Storage involves a complex pathway that initiates actin branching and blocks actin depolymerisation via a protein, adducin. But in competition, the musashi protein seems to stop the branching and destabilises actin polymers, causing memory loss. The balance of these processes is critical for retaining memories, but what controls which memories are kept and which lost isn’t known.

Addiction is one area where forgetting could be an advantage. ‘There are these associations that have been formed with drug use, so that addicts have hundreds of triggers that make them think about getting high and they experience craving and it can be very hard to resist,’ Miller explains. She started investigating drug memories in collaboration with her husband, Gavin Rumbaugh, also a Scripps neuroscientist. They previously discovered that molecular motor protein myosin II is involved in promoting actin polymerisation by physically pushing bundles of actin molecules around, and by blocking its action they could prevent memory consolidation.
Miller and Rumbaugh wondered if this might help diminish memories of methamphetamine addiction in rodents. They treated the amygdala region of rat and mouse brains with latrunculin, a natural product extracted from sponges that blocks myosin?II and actin polymerisation. They created associations between methamphetamine and a number of environmental cues and then tested the animals three days later to see if they sought out the drug when exposed to the same environment.6 ‘We got a really big surprise,’ Miller says. ‘When we gave them the drug just before testing them, the memory went away immediately. It didn’t make any sense to us because you can do the exact same manipulation in the exact same brain region with something like a fear memory, and it has no effect’.
After a great deal of head scratching, Miller and Raumbaugh concluded that different states of actin must exist in different types of memories. In some cases, such as in fear memories, polymerised actin is stable, but in the case of a pleasurable drug memory, actin at the synapse seems to continue to cycle with new G-actin monomers added at one end and removed at the other.
The existence of the memory is linked to the balance between making and breaking actin polymers – for polymer filaments to form, the rate of addition must be greater than the rate of removal. When latrunculin is added the actin polymer filaments fall apart, Miller explains ‘presumably because actin is continuing to cycle in the specific spines that are storing a drug-associated memory’. With fear memories, the actin cytoskeleton stabilises very quickly, so latrunculin has no effect.
Restoring memory

Understanding the mechanisms that make memories could help to treat neurodegenerative diseases such as Alzheimer’s, which the Alzheimer’s Society predict will affect one million people in the UK by 2021. Several approaches to stopping memory loss have been tried. One has been to target the transcription factor CREB (cyclic AMP response element binding protein), which activates genes needed for long term memory formation.
In 2003, Tim Tully, then at Cold Spring Harbor Laboratory in the US, showed that drugs could increase CREB activity and improve memory in mice genetically engineered with neurodegenerative disease and he continues to develop CREB-modulating drugs. Tsai says that several biotech companies are trying to develop histone deacetylse inhibitors for neurodegenerative diseases, but that there are big question marks around their safety. ‘The concern … is that they are not specific,’ she says: such drugs turn on and off other genes and therefore could have unforeseen side-effects.
Attention has recently turned to treatments that might improve the memory of people with Down’s syndrome, the condition caused by a third copy of chromosome 21. In 2007, Balance Therapeutics was set up to exploit work by Craig Heller and Craig Garner of Stanford University, US, and the company currently has a drug, pentylenetetrazole (PTZ), in clinical trials. Heller says the initial idea came from graduate student Fabien Fernandez, who suggested the problem was too much inhibition of memory mechanisms. ‘We tend to think of the brain as being like a puppet master pulling strings, but in reality the brain is more like the symphony conductor bringing some things up, quieting other things down,’ says Heller.
The Stanford team found that PTZ was able to increase the learning abilities of mice genetically engineered with Down’s syndrome7 by blocking receptors for the neurotransmitter GABA (gamma aminobutyric acid). These receptors open channels that allow chloride ions to flow into cells and counter the depolarisation process that sparks neuronal signalling. By dampening the overactive inhibition, memory can be improved.
Smart pills for all?
PTZ had no effect on normal mice, but other drugs certainly do and so-called ‘smart drugs’ are already in use. For example, modafinil, a drug licensed for treating narcolepsy, has become popular with students, who often obtain it online. It is thought to interact with a number of neurotransmitters and there is evidence it can enhance cognitive performance, including memory. Heller says he would be cautious in delivering cognitive enhancers to a general population, particularly given that there are already problems with the overuse of attention deficit drugs such as methylphenidate (Ritalin).
Safety issues aside, Anders Sandberg, bioethicist at the University of Oxford’s Future of Humanity Institute in the UK, thinks ‘there are no real knock-down arguments against enhancement’. He doesn’t believe there is a distinct line between treating cognitive deficits and enhancing normal performance. ‘There is always a lot of variability in human traits,’ he says. But many people are uncomfortable with the idea and argue that such drugs would give an unfair advantage to users, leading others to feel compelled to take them.
Sandberg thinks you need to consider whether an activity is solely competitive; for example, he says doping is ‘against the idea of what sport is about’. On the other hand, if we consider the primary purpose of university is to attain knowledge itself, rather than compete in exams, then memory enhancers might help maximise learning potential. Sandberg gives the example of a mathematician taking enhancers. ‘We don’t really care in what mindset he was when he figured out the clever theorem, we only care about the theorem.’ In jobs where people are routinely expected to perform for long hours, cognitive enhancers may also be a benefit. For example, a 2012 study on sleep-deprived surgeons showed they performed better in problem-solving tasks when taking modafinil.8
In the future, it is not so far-fetched to imagine taking memory-enhancing drugs along with your morning coffee, to stave off the effects of neurodegeneration and enhance your memory. While the sort of memory control suggested in science fiction may always be fantasy, we are certainly edging slowly closer to understanding and controlling what we remember and what we forget.
Rachel Brazil is a science writer based in London, UK
References
1 C A Miller and J D Sweatt, Neuron, 2008, 59, 1051 (DOI: 10.1016/j.neuron.2007.02.022)
2 C A Miller et al, Nat. Neurosci., 2010, 13, 664 (DOI: 10.1038/nn.2560)
3 A Fischer et al, Nature, 2007, 447, 178 (DOI: 10.1038/nature05772)
4 E Griggs et al, J. Neurosci., 2013, 33, 4 (DOI: 10.1523/jneurosci.2873-12.2013)
5 N Hadziselimovic et al, Cell, 2014, 156, 1153 (DOI: 10.1016/j.cell.2014.01.054)
6 E J Young et al, Biol. Psychiatry, 2014, 75, 96 (DOI: 10.1016/j.biopsych.2013.07.036)
7 D Colas et al, Br. J. Pharmacol., 2013, 169, 963 (DOI: 10.1111/bph.12169)
8 C Sugden et al, Ann. Surg., 2012, 255, 222 (DOI: 10.1097/SLA.0b013e3182306c99)











1 Reader's comment