
Claire Jarvis talks to the scientists trying to find new drugs to treat the inherited lung condition
There’s still no cure for cystic fibrosis. Despite improved treatments lengthening patient lifespans, a CF patient born between 2013–17 in the UK has an average life expectancy below 50. In the UK, around 10,500 people have cystic fibrosis, according to the Cystic Fibrosis Trust. Between 60–80% of people with cystic fibrosis suffer chronic respiratory infections, commonly caused by Pseudomonas aeruginosa or Staphylococcus aureus bacteria. Once the lungs are infected, the bacteria differentiate rapidly into sub-strains, develop resistance to antibiotics and become difficult to treat. Current antibiotic treatment strategies are equivalent to nuking the entire site from orbit with a battery of drugs, an approach which causes severe side effects, and in many cases the bacteria persist.
Chronic lung infection damages epithelial cells surfaces and leads to inflammation, impairing the lung’s ability to function. It’s estimated over 80% of cystic fibrosis patients are killed by these kinds of chronic lung infections. There’s a lot we don’t understand about how these bacteria colonise and spread within the lungs, and how bacteria–bacteria interactions affect their sensitivity to antibiotics.
Fortunately, scientists are finding new ways to model bacterial behaviour in infected lungs and gain insights into how infections take hold and resist treatment. These findings have the potential to help CF patients live longer and healthier lives.
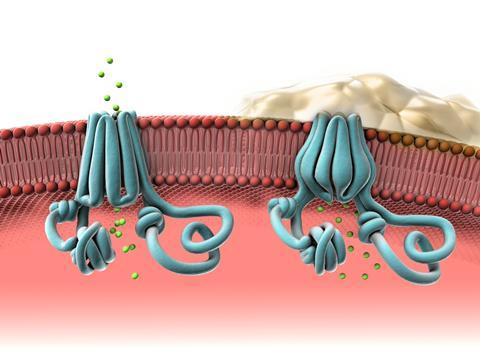
Cystic fibrosis is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene that affect the production of the CFTR protein. This protein forms ion channels in cell membranes throughout the body that regulate the flow of ions and water molecules in and out of cells. When the CFTR protein is not made correctly its ability to transport ions is compromised, affecting the balance of salt ions and water inside and outside of the cell. This imbalance leads to thick, sticky mucus in the lungs, pancreas and other organs. The mucus coughed up from the respiratory tract is called sputum.
Mucus that doesn’t clear is a breeding ground for bacteria and leads to these chronic lung infections in CF patients. It’s also hard for therapeutic agents to penetrate the mucus and eradicate the bacteria.
How many bugs?
The first step in understanding bacterial infections is to quantify what’s going on. The NIH Common Fund Human Microbiome Project (HMP) was established in 2008 to characterise healthy human microbiota and investigate their role in disease. The first phase of the project characterised oral, gut and skin microbiomes, but didn’t investigate the lungs. Culturing lung samples on a petri dish – the traditional way of identifying bacteria – didn’t show anything, so scientists concluded healthy lungs were sterile. Subsequent research with more advanced sequencing techniques showed that in fact multiple bacteria species inhabit healthy lungs.
However, the differences between a healthy and unhealthy lung microbiome is ‘a pretty controversial area right now’, explains Ann Field, senior director of drug discovery at the Cystic Fibrosis Foundation, a US non-profit. Some studies identify lists of organisms that appear to be present in lungs where there isn’t an infection, but other studies identified considerable overlap in the types and quantities of organisms in healthy and infected lungs. ‘The jury is still out on whether there is a “healthy lung microbiome”,’ Field says.
The most common method of quantifying bacteria is with 16S ribosomal RNA (rRNA) gene sequencing, where PCR amplifies the 16S rRNA gene present in DNA collected from sputum samples. The 16S rRNA gene is present in all bacteria, and acts as a barcode for different species.
When they conducted 16S rRNA sequencing on sputum from 77 cystic fibrosis patients, researchers at Emory University and Georgia Institute of Technology, both in Atlanta, US, found the presence of Pseudomonas, Staphylococcus and Achromobacter in significant quantities correlates to reduced lung function in CF patients, while increased microbiome diversity correlates to improved lung function. However they also found exceptions to these trends.
There are some drawbacks to using DNA-based methods to quantify bacteria in the lungs. In CF patients a lot of host and bacterial DNA is present in samples, but the quantities of DNA may not represent the actual composition of bacteria present. ‘Bacterial DNA accumulates at different rates with different stabilities; a lot of bugs in the airways make pretty good DNAases, so they tend to degrade the DNA around them. Whereas with pathogens like Pseudomonas the DNA accumulates continually,’ says Michael Surrette, a professor of microbiology at McMaster University in Ontario, Canada.
Surrette argues a more accurate way to quantify bacteria is to look for bacterial RNA. The drawback of RNA-based sequencing methods is although it gives a more accurate snapshot of species present, RNA isn’t stable, and has half-life of minutes. Extracting RNA from sputum is also difficult. ‘Sputum is the worst possible sample to do this with,’ Surette says. His group has developed a protocol that breaks down the saliva matrices, stabilises the RNA before amplification and sequencing, and takes less than five minutes to perform.
Antibiotic resistance
More than 50% of adults with CF in the US are infected with Pseudomonas, of which approximately one-third is multidrug-resistant. Pseudomonas is an opportunistic pathogen that only infects people with weakened immune systems. There are multiple mechanistic pathways through which Pseudomonas acquires antibiotic resistance. Pseudomonas’ habit of forming protective biofilms adds to the difficulty of eradication.
To study resistance, Steve Diggle’s team at Georgia Institute of Technology grew Pseudomonas biofilms on plastic beads with synthetic sputum media over the course of 50 days. They found that within a month Pseudomonas had enough mutations in the IasR gene – which promotes the creation of virulence proteins – to gain tolerance to several beta-lactam antibiotics, even when the samples weren’t treated with antibiotics.
Diggle’s lab also showed that more aggressive strains of Pseudomonas release pyocins that kill competing Pseudomonas strains. This ensures the most virulent – and deadly – strains come to dominate CF patient’s lungs.
I use the example of galaxies in the universe: there’s a lot of them but most are too far apart from each other to interact
Another complicating factor is that the presence of multiple microbes in the lung can affect their ability to resist antibiotics. A strain of bacteria that was cultured and purified from a patient sample might show sensitivity to a particular antibiotic in the dish, but appear resistant to that treatment within the context of a polymicrobial infection. ‘The implication is that these microbes are interacting in some way which changes their biology and physiology such that they’re not sensitive to the antibiotic,’ says George O’Toole, a microbiologist at Dartmouth College in New Hampshire, US.
O’Toole’s lab looks for the cross-feeding relationships within polymicrobial communities. The waste metabolite of one bacterial species can be food for another. Using computer simulations with a known selection of bacteria and pre-existing nutrients, his team can model the exchange and accumulation of metabolites in polymicrobial communities. They found that Pseudomonas often grew to become the dominant bacteria, but the presence of rarer pathogens that also consumed the same food metabolites as Pseudomonas could impede its growth. How these kinds of microbial interactions influence antibiotic sensitivity is not yet known.
Not all scientists agree that a lung microbiome where multiple microbes interact with each other exists. ‘I use the example of galaxies in the universe: there’s a lot of them but most are too far apart from each other to interact,’ says Diggle, who thinks the same could be true of bacterial colonies in the lung. He argues that seeing interactions between two bacteria species in a dish doesn’t mean they normally interact with each other in the lung.
‘Essentially every drug discovery program is run looking at the ability of an antibiotic to treat a single microbe in a culture,’ says O’Toole, who doesn’t see that as an effective approach in CF when nearly every patient has polymicrobial infections. ‘We need to think about a drug discovery pipeline where you’re actually looking for drugs that will be effective in the context of a mixed infection.’
Modelling CF lungs
Another challenge when it comes to cystic fibrosis is effectively modelling the disease and microenvironment within unhealthy lungs. Lungs are difficult to image and analyse when they’re still in use. When sputum is coughed up for analysis, clinicians don’t know what part of the lungs the sample came from, or whether it’s a mix of bacterial colonies from multiple locations. Without effective models, scientists cannot effectively study the disease and develop better treatments.
‘Unlike human CF patients, CF mice fail to develop spontaneous lung disease or chronic bacterial infections. 2D cell culture models also do not encompass many of the mechanisms and treatment responses of respiratory diseases,’ says Sean Murphy, a bioengineer at Wake Forest Institute for Regenerative Medicine in South Carolina, US.

Performing real-time experiments on living lungs is almost impossible, so some researchers have created miniaturised organs to study. Researchers at Harvard University in Massachusetts, US, have spent many years building organs-on-a-chip devices that mimic physiological conditions. Their organ chips are microfluidic culture devices that have two hollow parallel channels that are less than a millimetre wide. They are separated by a porous membrane that is coated with an extracellular matrix to mediate cell adhesion in the body and in vitro actions. ‘They’re the size of a computer memory stick, a little thumb drive,’ says Donald Ingber, founding director of the Wyss Institute for Biologically Inspired Engineering at Harvard. The results are living, 3D cross-sections of major functional units of the organs. They have individual lung chips that stand in for the alveoli, air sacs and airways.
The Wyss Institute’s lung on-a-chip project began in 2018 with a grant from the Cystic Fibrosis Foundation. When researchers put lung endothelial cells from CF patients onto chips the mini organs produced thicker mucuses, and had abnormal capillary motion and a greater inflammatory response. Ingber says they will use these chips to reverse engineer the disease process by changing physiological variables.
There are hopes that lungs on a chip can help explain the mechanism of CF lung inflammation. ‘There is some evidence that the CF inflammatory responses are either exaggerated or suppressed independently of CF lung disease,’ notes Murphy, even though chronic bacterial infections and damage also have a significant impact on the inflammatory response. His team has built their own lungs-on-a-chip system that can use different combinations of CF or non-CF immune cells and epithelium to better understand mechanisms of inflammation in the CF lung and immune system.
Modulators
Despite many unanswered questions about the underlying biology of the disease and its accompanying chronic infections, the treatment and management of cystic fibrosis is undergoing a massive paradigm shift with the recent introduction of CFTR modulator drugs. They are called modulators because they modulate the function of the CFTR protein so that it can serve its primary function: to create a channel for chloride ions to flow across the cell surface.
When proper chloride flow is reestablished, mucus becomes rehydrated inside the lungs and other organs. Although current modulator drugs don’t completely restore proper chloride flow, they can improve the flow enough to relieve symptoms for people with CF. Symkevi – a drug from Vertex Pharmaceuticals – is a combination of modulator drugs ivacaftor and tezacaftor was approved in December 2018 for use in the EU. Vertex followed this with Trikafta – a treatment with a third modulator drugs elexacaftor – which was approved by the FDA in October 2019. The European Medicines Agency is still reviewing the application for authorisation, and will make a decision at the end of 2020.
We can’t afford to let our guard down
Trikafta is approved for patients aged 12 and older who have at least one F508del mutation in the CFTR gene, which is about 90% of cystic fibrosis patients. In clinical trials, people with two copies of the F508del mutation had a 3.4% increase in lung function after taking Symkevi compared to treatment with the placebo group. People who took Trifkafa had an additional 10% increase in lung function compared to patients taking Symkevi.
In addition to modulators there are several classes of therapies targeting different aspects of the CFTR protein, although these have yet to be approved for widespread use. Potentiators are CFTR modulators that hold the gate open so chloride can flow through the cell membrane. Correctors help the CFTR protein to form the right 3D shape so that it is able to traffic ions to the cell surface. Finally, CFTR amplifiers increase the number of CFTR proteins on the cell surface. If the cell makes more CFTR protein, it increases the flow of chloride ions across the cell membrane. Amplifiers, which are being developed and tested, are not yet available commercially.
Despite the optimism surrounding improved outcomes for CF patients with the arrival of modulator drugs, Surrette cautioned that modulators may lead to a new set of infection issues. Even when these modulators clear up the mucus in adult CF patient lungs, scarring from past infections and exacerbations will still remain, which means CF lungs will never quite look like healthy lungs. Researchers aren’t sure if patients on CFTR modulators will gain a permanently healthy lung microbiome, or if pathogens will eventually return. ‘This is going to be an environment for potential new pathogens that we haven’t seen before. We can’t afford to let our guard down,’ says Surrette.
Claire Jarvis is a science writer based in Atlanta, US











1 Reader's comment