
Pairs of frustrated radical species can selectively activate carbon–hydrogen bonds that were previously inaccessible with existing methods. The reagents are easily tuned, allowing them to target different C–H bonds on substrate molecules as required.
Reactions that activate aliphatic C–H bonds are very useful in organic chemistry, but the stability of these ubiquitous bonds makes selective functionalisation particularly challenging. Current strategies include transition metal catalysis and enzymatic methods, but these approaches tend to have limited functional group tolerance and often only target the most reactive bonds.
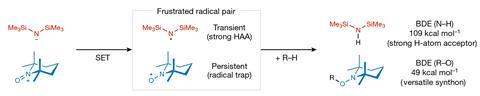
An alternative strategy uses frustrated Lewis pairs – bulky acid and base pairs which are unable to quench themselves due to steric hindrance – which are known to effectively activate strong covalent bonds in small molecules. More recently, researchers have extended this study to frustrated radical pairs (FRPs), which contain two bulky single-electron species that are prevented from combining with and annihilating each other.
The identity of each component of the radical pair is crucial for forming an effective reagent. ‘Firstly, they should be relatively bulky so they don’t react with each other and secondly, the electron transfer to form them must be quick,’ explains Song Lin an organic electrochemist from Cornell University, US. ‘You also need to form a pair of radicals that will be useful in synthesis, two species that play different roles in the bond activation.’
With these considerations in mind, Lin’s team generated an FRP using a hexamethyldisilazide anion (HMDS-) and an N-oxoammonium cation (TEMPO+) as precursors. Importantly, the resulting radicals possess orthogonal reactivity. HMDS・ is a transient radical – an extremely reactive species which acts as a strong hydrogen atom abstractor (HAA) – while TEMPO・ is a persistent radical, acting as a radical trap for any single-electron species left in solution. In the presence of a C–H bond, the transient HMDS・ rips off the hydrogen atom, leaving an alkyl radical which is subsequently mopped up by the persistent TEMPO・ radical trap. The product TEMPO adducts are then readily cleaved and derivatised into a diverse panel of other functional groups.
The team demonstrated broad substrate scope across over 40 examples, showing tolerance to a range of functional groups including halides, nitriles, methyl esters and heteroarenes. ‘Organic radicals are very interesting – they’re highly reactive, but they’re very selective as well,’ explains Lin. ‘These FRPs provide complementary functional group compatibility to transition metal catalysis.’
Rebecca Melen, a catalysis researcher from Cardiff University in the UK is particularly impressed by this aspect of the work. ‘The functional group tolerance and selectivity for a particular C–H bond is excellent,’ she says. ‘Interestingly, they’ve also demonstrated that by changing the HAA species, you can change the regioselectivity of the reaction.’
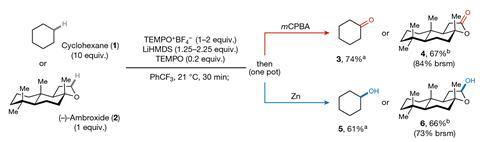
Owing to the bulky nature of FRPs, hydrogen abstraction generally occurs at the less hindered (and typically less reactive) primary C–H bonds. The team investigated whether this steric effect could be used to control the regioselectivity of the reaction and varied the hydrogen abstracting species, employing bulkier hexaphenyldisilazide radicals (HPDS・) and smaller tert-butoxide radicals for comparison. Across 12 substrates, the smaller hydrogen abstractor targeted the more reactive but crowded tertiary positions, whilst bulkier HPDS・ only interacted with the less-activated but sterically more-accessible sites.
‘Overall, this is an outstanding piece of work and provides a significant advance in the selective functionalisation of inert C–H bonds,’ says Melen. ‘The next steps should be to find out what other reactions can be performed using this and other FRP systems.’
Lin’s team are currently working on improving this methodology but ultimately hope that FRPs could be used in reactions beyond just C–H activation. ‘One limitation of the method right now is that we do use a strong base, so in the short term we want to develop milder second-generation systems,’ he says. ‘Long term, we want to see how we can leverage this idea to enable the activation of other types of chemical bonds.’
References
Z Lu et al., Nature, 2023, DOI: 10.1038/s41586-023-06131-3












No comments yet