The enzymes all contain iron atoms and the most effective also boast molybdenum or vanadium. Chemists have spent decades debating which of these metal atoms is responsible for binding and reducing dinitrogen, and have made various analogue complexes to compare with the natural catalyst.
Jonas Peters at the California Institute of Technology, US, has now developed the first iron-based catalyst that can reduce dinitrogen to ammonia in solution,2 which could prove to be a useful model for nitrogenase. ‘Lots of folks have been working to achieve this for many years,’ says Brian Hoffman at Northwestern University, US. ‘Jonas' achievement is really terrific.’
Although previous iron complexes have been able to crack open dinitrogen,3 none have worked catalytically. In contrast, two of the leading modelcatalysts rely on molybdenum. 4,5 This has bolstered the idea that dintrogen is reduced at the molybdenum atom in a segment of nitrogenase called the iron–molybdenum cofactor (FeMo-co).6 Peters’ complex is a significant development because it ‘levels the playing field’, says Hoffman.
Others are not convinced. ‘It is a wonderful achievement, but nothing has been proven about nitrogenases,’ says Richard Schrock at the Massachusetts Institute of Technology, US, who created one of the molybdenum model catalysts and still considers molybdenum to be the most likely nitrogenase reaction centre. ‘His complex is not a model for nitrogenase, except to the extent that ammonia is produced.’
Fast but vulnerable
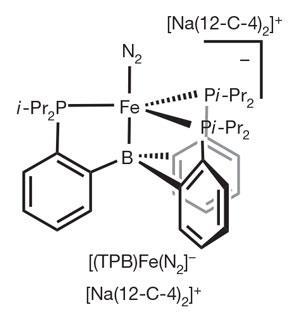
The catalyst contains a single iron atom bound to a tris(phosphine) borane ligand, and primed with a molecule of dinitrogen (see image). Electrons from the strong reducing agent potassium graphite and protons from the boric acid HB(3,5-(CF3)2-C6H3)4, also known as HBArF4, reduce the dinitrogen to ammonia. Remarkably, only about half of the protons and electrons in the reaction mixture are lost as hydrogen gas, suggesting that the catalyst acts quickly.
But it is also vulnerable: each molecule of catalyst can make about seven molecules of ammonia before it stops working. Still, even the best molybdenum catalysts typically only manage a dozen catalytic cycles before conking out, says Peters.We’re not trying to replace the Haber-Bosch process, that chemistry works very well
He is now working with Hoffman to compare the catalyst with the FeMo-co. Techniques such as electron nuclear double resonance (ENDOR) spectroscopy can help to reveal the shape and electronic structure of metal complexes. If ENDOR data on Peters’ complex matches those from intermediates in the nitrogenase catalysis cycle, it may reveal how electrons and atoms shift around the FeMo-co. ‘The goal is to correlate the two,’ says Hoffman.
These studies could also help to determine the role of a carbon atom that lies at the core of the FeMo-co. This carbon atom, only discovered in 2011, 7 might be helping to fine tune the activity of the catalytic iron centre in some way, suggests Peters. His team has already created versions of their complex containing a carbon atom bonded to iron, rather than boron. These can also reduce dinitrogen to ammonia and may offer a better model for the FeMo-co. In unpublished studies, Peters has isolated various catalytic intermediates and measured how the length of the iron–carbon bond changes from step-to-step.
While the research could also prompt fresh approaches to breaking some of the toughest bonds in chemistry, these complexes are certainly not about to displace the highly-efficient Haber–Bosch process for making ammonia on an industrial scale, says Peters. ‘One should be very clear about how useful Haber–Bosch is,’ he says. ‘We’re not trying to replace it, that chemistry works very well.’
References
1 W A Bulen and J R LeComte, Proc. Natl. Acad. Sci. USA, 1966, 56, 979
2 J S Anderson, J Rittle and J C Peters, Nature, 2013, 501, 84 (DOI: 10.1038/nature12435)
3 M Rodriguez et al, Science, 2011, 334, 780 (DOI: 10.1126/science.1211906)
4 D Yandulov and R Schrock, Science, 2003, 301, 76 (DOI: 10.1126/science.1085326)
5 K Arashiba, Y Miyake and Y Nishibayashi, Nat. Chem., 2011, 3, 120 (DOI: 10.1038/nchem.906)
6 R Schrock, Nature Chemistry, 2011, 3, 95 (DOI: 10.1038/nchem.977)
7 K Lancaster et al, Science, 2011, 334, 974 (DOI: 10.1126/science.1206445)







No comments yet