Study reveals that structural transformations occur just as fast as molecular vibrations
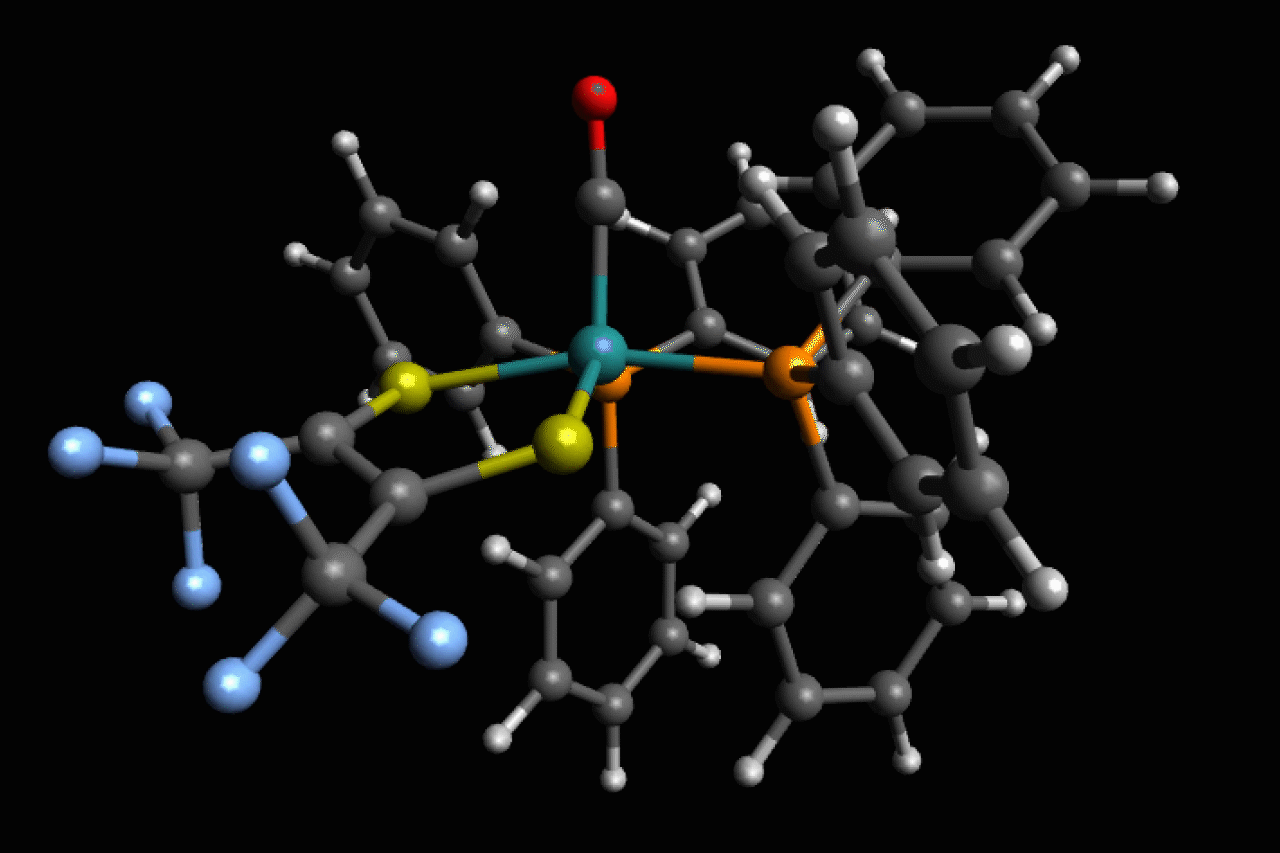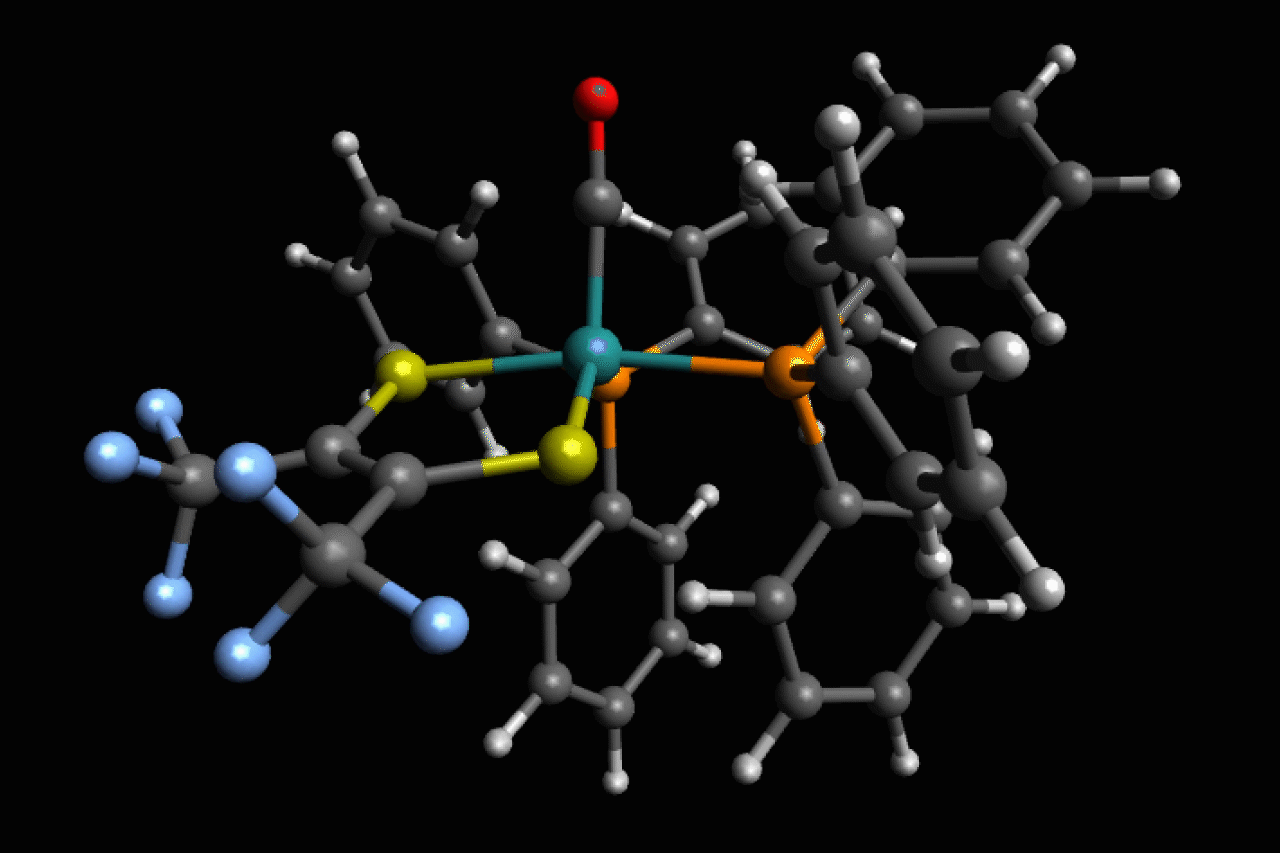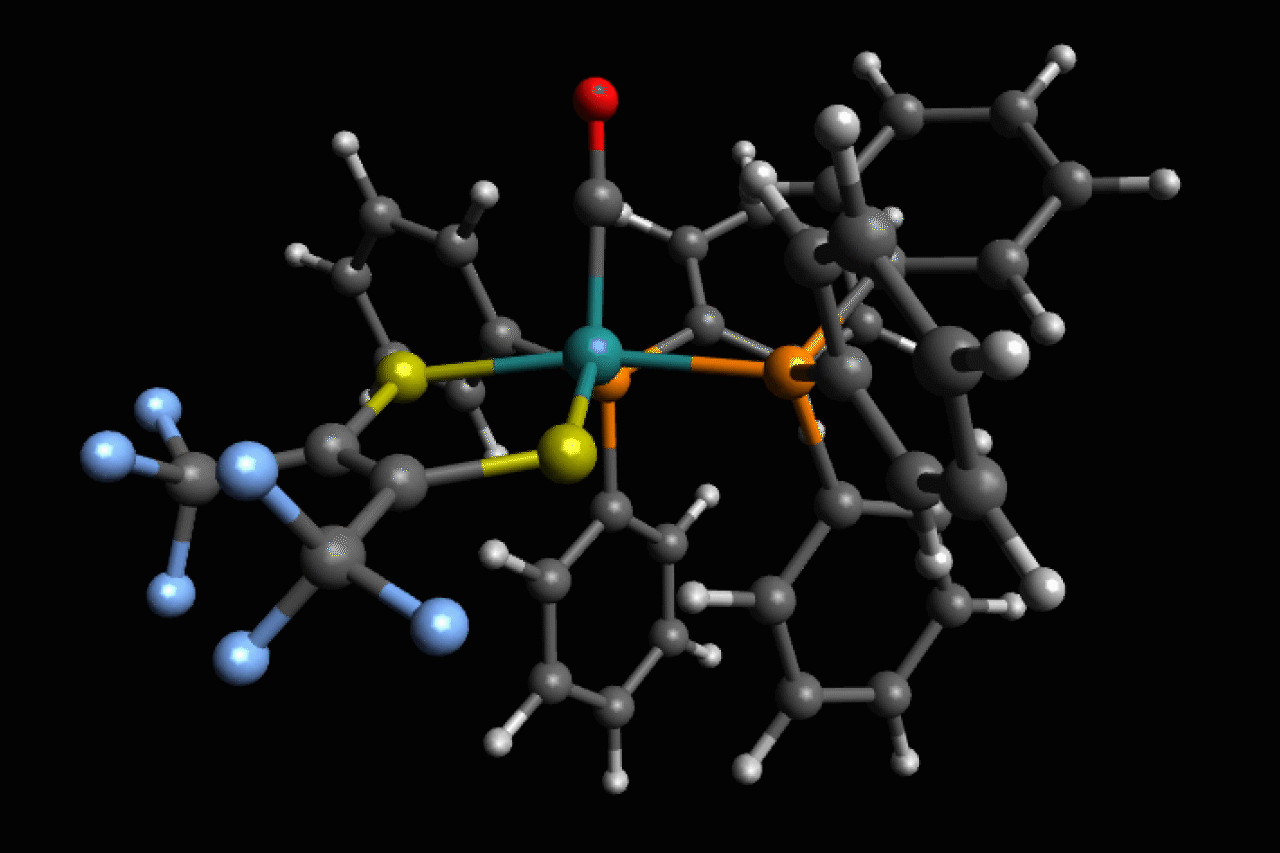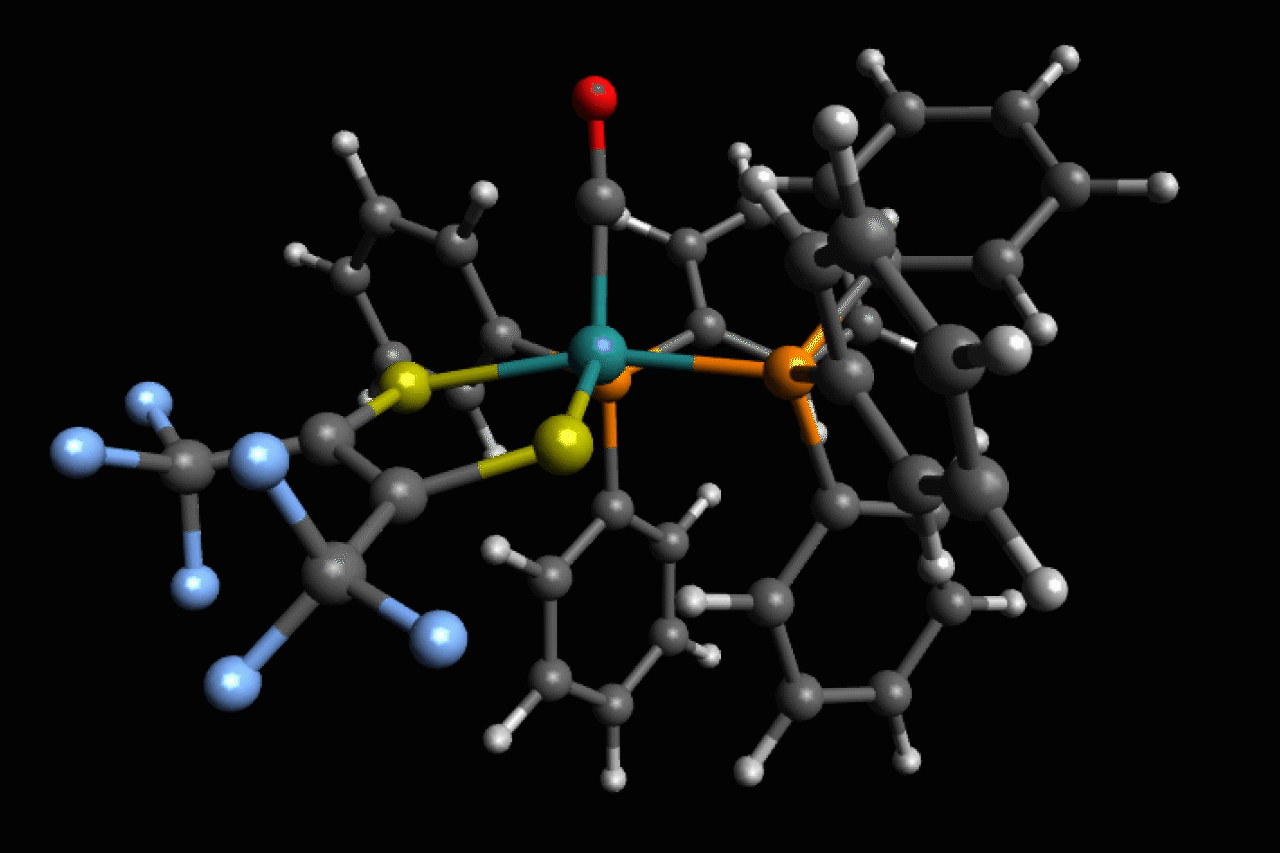
Scientists in the US have observed ultrafast structural isomerisations at picosecond (1 × 10-12s) speeds, adding another chapter to the story of Berry pseudorotation in the process.1
Structural changes and transformations in the geometries of molecules are fundamental to how they interact with each other. However, the speed at which molecular shapeshifting can occur poses problems for scientists who study it. The dynamics of these chemical exchanges are typically investigated using techniques like nuclear magnetic resonance (NMR) spectroscopy, however NMR can only observe processes that occur in the millli- to microsecond timescale – anything faster is tricky to identify.
Ru(S2C2(CF3)2)(CO)(PPh3)2, a five-coordinate ruthenium complex, was first reported in the 1970’s by Joel Miller and Alan Balch, at the University of California, Davis, who noticed that, depending on the solvent used, both orange and violet crystals formed.2 Analysis of these crystal structures revealed two different isomers, but in most spectroscopic techniques they are averaged and indistinguishable as the transition between the two is happening so fast.
Now, using ultrafast two-dimensional infrared (2D IR) spectroscopy, Clifford Kubiak, from the University of California San Diego, and colleagues have directly observed the rapid structural isomerisation of Ru(S2C2(CF3)2)(CO)(PPh3)2. 2D IR spectroscopy uses three ultrafast infrared pulses to study molecular structures and dynamic processes at the sub picosecond timescale. For context of how fast a picosecond is, light (travelling at 299,792,458m/s) only travels ~0.3mm in a picosecond. The first two pulses identify and activate vibrations within the molecule, and the third pulse probes how these vibrations change.
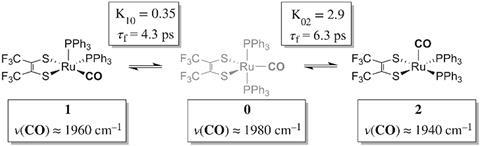
‘With 2D IR we can follow the frequency evolution of a molecule, which is basically the structural evolution of the molecule with respect to time,’ summarises Wei Xiong, who led the 2D IR spectroscopic aspect of the study. ‘Linear spectroscopy is like having a camera and taking one long exposure shot – if the object moves around, your picture gets blurred. 2D IR is like using a camera to take snapshots, each time very fast. In the first snapshot you capture a molecule’s current state, which has a certain absorption frequency. If you then take another snapshot and the molecule has changed, then its characteristic absorption frequency will have also changed.’
The big picture
Potentially the most interesting conclusion of this work could be the connection between molecular vibrations and structural isomerisation, comments Kubiak: ‘It shows that these rather elaborate structural rearrangements of large inorganic or organometallic molecules can be very, very fast, and only a little bit slower than the periods of molecular vibrations themselves. So we’ve concluded that the reason these are so fast is that the motions that cause the molecules to rearrange are on the same trajectory as the molecular vibrations.’
‘The rearrangement is simply following motions that are naturally occurring in the molecule. If those motions exceed the normal vibrations, you end up with a different structural isomer. This can happen with just a couple of shakes of the molecule,’ concludes Kubiak.
Here, Ru(S2C2(CF3)2)(CO)(PPh3)2 undergoes what is known as Berry pseudorotation, so named after Stephen Berry who first described the process in 1960.3 Molecules with trigonal bipyramidal (such as PF5) or square pyramidal (ie ClF5) geometries have both apical (at the apex) and equatorial ligands. If a molecule has enough energy, the apical ligands can close together like a pair of scissors, however to maintain stability this motion pushes any equatorial ligands further apart.
Stephen Berry, now at the University of Chicago, comments: ‘The isomerisations analysed in this work constitute an elegant advance in the use of ultrafast spectroscopic methods to elucidate the pathways of molecular rearrangements. To extract the thermodynamic parameters, the shape of the multi-minimum potential surface and the corresponding geometric structures of the isomeric forms of the complex, the researchers have used infrared pulse spectroscopy in a strikingly powerful way.’
However, as if often the case with scientific discoveries, these results pose further questions, concludes Berry. ‘The work suggests a possible next step: if the reaction were to be carried out in an optically-active solvent, one might be able to determine which of the PPh3 groups is favoured to move between an equatorial and apical position. Would a chiral environment favour clockwise or counterclockwise pseudorotation?’
Berry pseudorotation is revelant across the chemical sciences. ‘In the 1960s, Berry pseudorotation had helped explain the detailed mechanism of phosphate ester hydrolysis through a trigonal bipyramid intermediate and biological relevance in explaining mechanistic details of enzymatic catalysis by ribonuclease,’ comments Edward Dennis, an expert in phosphate biochemistry at the University of California San Diego. ‘The current work suggests that the equatorial–apical preferences between CO and phosphine ligands is extremely small, but sufficient that two different stable crystal forms were isolatable and explainable by small barriers to Berry pseudorotation.’
References
1 T M Porter et al, Chem. Sci., 2018, DOI: 10.1039/c8sc03258k (This article is open access.)
2 A L Balch and J Miller, Inorg. Chem., 1971, 10, 1410 (DOI: 10.1021/ic50101a019)
3 R S Berry, J. Chem. Phys., 1960, 32, 933, DOI: 10.1063/1.1730820










No comments yet