Conophylline
Conophylline
Named reactions and reagents are both a blessing and a curse to the synthetic organic chemist. Most scientists with even a basic grasp of organic chemistry will know at least one or two (the Wittig and Grignard reactions are probably top of the pile), but even the largest tome can only describe a subset of the full gamut. As a result, the well-known names become a lingua franca in synthesis, while the ’long-tail’ of more obscure concoctions develops into an exercise in literature hell.
This introduction is actually a partial excuse for the fact that I’ve never heard of the Polonovski-Potier reaction, the keystone of a remarkable synthesis by a team led by Tohru Fukuyama at the University of Tokyo, Japan.1 And that excuse is made necessary as this reaction really isn’t all that obscure at all - it even features in my favourite guide to named reactions!2
Aside from its compelling synthetic challenge, conophylline is also a rather potent biological agent, showing potential for treatment of type 1 diabetes, as well as a possible therapeutic role in pancreatic cancer.3,4 However, my interest (like the authors’) is focused on their application of the Polonovski-Potier reaction.
Key to the group’s synthetic strategy was recognising that conophylline is actually a pseudo-dimer, so they could use the same strategy to make both halves. This firstly involved building a rather complex indole unit, using typical aromatic chemistry for the majority of the functionality, saving a rather neat Stille coupling to attach a methyl acrylate moiety. With two planar rings installed, the more complex 5,6,5-tricyclic system was next, but this was something the team did with apparent ease.
Stereochemistry was provided by a functionalised dihydrofuran derivative, containing a key protected primary amine side-chain. Using another more familiar named reaction (the Mitsunubu coupling), these two advanced intermediates were united through the protected amine (figure 1). Deprotection then prompted the furan to rearrange to a tetrahydropyridine, featuring a strategically placed olefin.
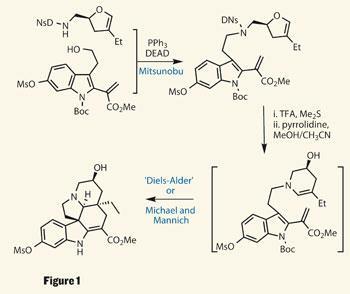
Name that reaction
The reason for all the unsaturation in the molecule was then revealed: an intramolecular cyclisation that appears to be a Diels-Alder type cycloaddition. However, Fukuyama explains that this is more likely a stepwise process. The enamine undergoes Michael addition to the acrylate moiety, followed by a Mannich reaction of the indole with the newly formed iminium ion to give the product as a single isomer. Elimination of the hydroxyl, now surplus to requirements, reintroduced an alkene, which was immediately oxidised to an epoxide.
By exactly the same strategy, the group made a second similar pentacyclic fragment, differing only in the functionalisation around the indole. Further oxidation using m -chloroperbenzoic acid - another favourite in the synthetic world - then oxidised not the remaining alkene, but the amine. This provided an amine oxide, and set the stage for the Polonovski-Potier reaction, in which elimination of water from the amine-oxide generates an iminium ion, to which a nucleophile can add (figure 2).
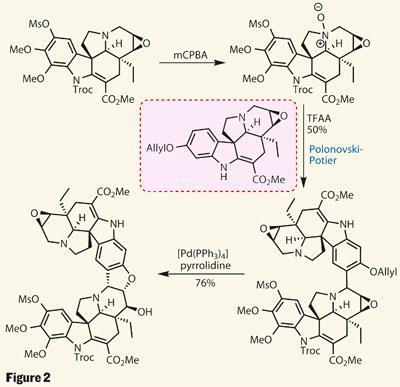
In this case, the nucleophile is the indole ring of the first fragment. Combining the two halves with a little acid resulted in an exceptional yield of the coupled product, as a single isomer. A palladium catalyst then selectively removed the allyl protecting group from the phenol, which attacked the nearby epoxide to complete the final ring.
Removing the two remaining protecting groups in base finished the synthesis, completing a hugely complex molecule in remarkably few steps.
Paul Docherty is a science writer and blogger based in Reading, UK
References
1 Y Han-ya, H Tokuyama and T Fukuyama, Angew. Chem. Int. Ed., 2011, DOI: 10.1002/anie.201100981
2 L Kürti and B Czakó, Strategic applications of named reactions in organic synthesis, Focal Press, 2005, p 355
3 T-S Kam et al, Tetrahedron Lett., 1992, 33, 969 (DOI: 10.1016/S0040-4039(00)91590-9)
4 K Umezawa et al, Anticancer Res., 1994, 14, 2413












No comments yet