BRSM gets to the core of a divergent synthesis of this natural product family
In 2012, Erick Carreira’s group from the Swiss Federal Institute of Technology (ETH) in Zurich reported the total synthesis of indoxamycin B.1 This 24-step organometallic tour de force resulted in a structural reassignment and set the bar rather high for future work on this family.
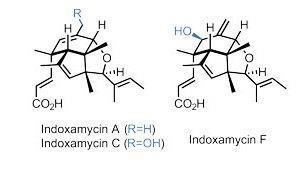
However, given their alluring dense functionalisation, moderate biological activity and several hitherto unsynthesised members – and with the prize of ‘first asymmetric synthesis’ still up for grabs – it seemed unlikely that the synthetic community was finished with the indoxamycins.
I’m sure many chemists aren’t too surprised to see their rather unusual structures reappear in the literature. That said, if I’d had to bet on who would be the next to publish in this area, I’m fairly certain that Hanfeng Ding’s group at Zhejiang University in China would not have been top of my list. In fact, this route to all the remaining members of family appears to be the team’s first independent total synthesis. In an impressive paper, they complete racemic syntheses of indoxamycins A, C and F; reassign their structures (in line with Carreira’s correction of indoxamycin B); and develop a new palladium-catalysed reaction to render their route enantioselective.2
Targeting a group of several similar molecules – whether natural products or analogues for medicinal chemistry – requires different tactics to those used to prepare a single molecule. A chemist considering only a single target will usually devise a convergent synthesis, to maximise the overall yield (and brag about how short the longest linear step sequence is). To make a group of similar molecules, a divergent strategy is usually more desirable.
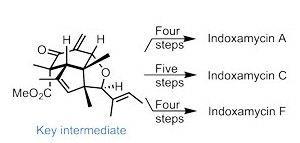
This typically involves designing an efficient route to an advanced intermediate, from which all the targets can be reached in as few steps as possible, minimising redundant operations. This tactic is skillfully employed by Ding’s team, identifying a compound containing all the shared stereocentres of the indoxamycins (figure 1). They construct this rather intricate tricyclic core using two very powerful transformations, leaving just one stereocentre to set – the one not conserved among family members.
The first of these is a remarkable palladium-catalysed reductive 1,6-enyne cyclisation (figure 2), in which an alkyne is coupled to one side of a racemic dienone. This simultaneously establishes three contiguous stereocentres with great control. Using an achiral catalyst gives the enone product in an excellent 87% yield, giving a great platform to proceed to the racemic natural products. It also gives a single diastereomer – meaning the alkyne selectively couples to the side of the dienone that gives a syn arrangement of the two methyl groups.
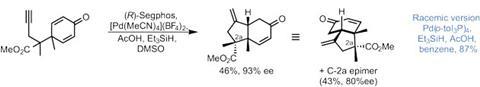
The team currently can’t explain this diastereoselectivity. However, that hasn’t stopped them developing a new asymmetric version to fish out the desired enantiomer of the product (figure 2). With the right combination of palladium source and chiral ligand, the catalyst overrides the natural selectivity, and the reaction becomes stereodivergent: each enantiomer of the racemic starting material is converted to a different enantioenriched product.
The final tetrahydrofuran ring is formed by an efficient cascade (figure 3), beginning with substrate-controlled 1,2-addition of a Grignard reagent to the aldehyde. The resultant alkoxide immediately undergoes 1,4-addition into the nearby enone, generating an enolate, which is quenched with
N,N-dimethylmethyleneiminium chloride to form an alkene.
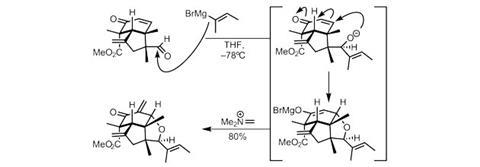
Isomerising this exocyclic double bond under acidic conditions gives the key intermediate that can be rapidly converted to indoxamycins A, C and F as planned.
One more interesting reaction occurred en route to indoxamycin F: reducing the ketone gives the wrong configuration of the secondary allylic alcohol, making it necessary to invert this stereocentre. However, instead of using standard Mitsunobu inversion chemistry, the group employs a double sigmatropic rearrangement to move the hydroxyl group elsewhere in the molecule – then return it to its original position but on the opposite face to where it started. A fitting end to a route full of creative solutions; I look forward to future work from the Ding group!
BRSM (@BRSM_blog) is a postdoctoral researcher and blogger based in the US











No comments yet