Even though 14 years have passed since those days of being entirely terrified by frontier molecular orbital theory, the capacity for nature to astonish me with architectural ingenuity has remained undimmed. One glance at the kingianin family of natural products did this again – the remarkable and unusual cyclobutane functionality kindling thoughts of ‘how the hell..?’
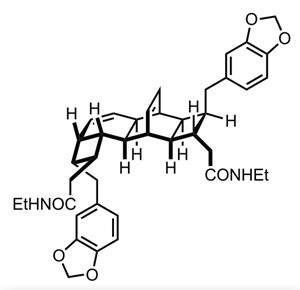
Of course, I’m not the only chemist to think these thoughts, and thankfully many others have suggested very reasonable biosynthetic processes to account for the kingianins’ existence. One likely proposal is dimerisation of a somewhat less complex bicyclo[4.2.0]octadiene system via a Diels–Alder [4+2] cycloaddition (figure 1), but this still leaves the 6,4-fused cyclobutane problem unanswered.1
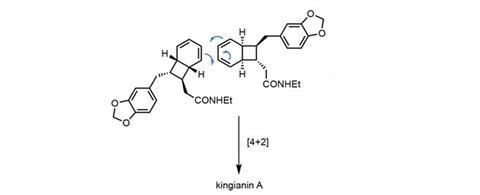
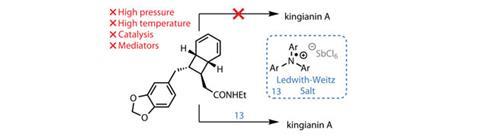
Stepping forward with a bold but reasonable alternative is a team led by Michael Sherburn of the Australian National University, Canberra.3 However, they firstly needed to build the exotic bicyclo[4.2.0]octadiene precursor – and for this, they trusted Black.
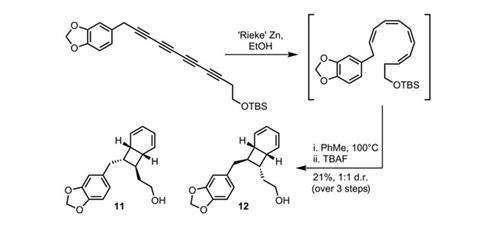
With the acyclic tetraene intermediate in mind, the team work towards a conjugated tetrayne analogue. The advantage of this route over building the tetraene system directly (through sp2–sp2 couplings) was speed and reaction efficiency – indeed, synthesis of the diyne precursors required only two and three steps respectively. Directly coupling the trimethylsilyl-protected diyne fragments was also quick, but somewhat less efficient. The suboptimal yield from this reaction can perhaps be attributed to the lack of differentiation between the partners, and consequent competing homo-dimerisation reactions.
However, completing such a complex intermediate in four steps is reward enough, as this allows the team to progress quickly to a tricky stereoselective reduction (figure 2). Using a highly activated form of zinc (named after its developer, Reuben Rieke) and ethanol as a source of hydrogen, they produce the desired bicyclo[4.2.0]octadiene target, but as a mixture of diastereoisomers. The yield may be relatively low, but again the efficiency in step-count more than makes up for this.
Paul Docherty is a science writer based in Reading, UK











No comments yet