Combining photocatalysis with organocatalysis opens doors to chiral quaternary centres
In recent years, the advent or renaissance of photochemistry – personal experience dependant – has been glaringly clear. Photoredox chemistry, which typically uses visible light in combination with a light-absorbing catalyst to mediate redox processes, has led to a multitude of sleek publications that have dominated the field. That said, impressive recent contributions in ‘classical’ photochemical reactions – using ultraviolet radiation to mediate [2+2] cycloadditions of alkenes or the Paternò-Büchi reaction with carbonyls – are also refocusing attention on a field that could be said to have lost its shine.
Classical photochemistry enables reactions that would be ‘forbidden’ under thermal conditions. It can therefore provide access to biologically active scaffolds not easily prepared using traditional chemistry, bringing contemporary applications to a field with an enthralling history of fundamental studies. In the early days of photoredox catalysis, around 2009–2010, dissenting voices dismissed it as a fad, but continued excellence in the field and exponential growth of applications is showing it to be anything but. Novel transformations using widely available reagents such as alcohols and arylnitriles have brought down the curtain on the critics. These unorthodox reactive substrates can be used to generate synthetically tractable radical intermediates under ambient conditions.
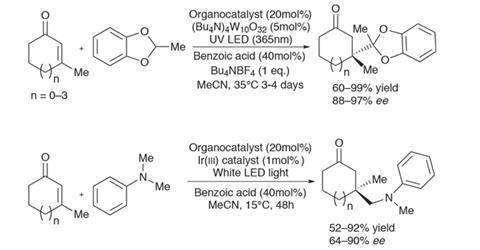
Paolo Melchiorre and his team at the Institute of Chemical Research of Catalonia, Spain, are pushing the boundaries of photoredox catalysis into the realm of classical organometallic conjugate addition reactions. The team has discovered an asymmetric way to form quaternary carbon centres by adding nucleophilic radicals to enones. Using a cleverly conceived redox-active organocatalyst, in combination with an appropriate photocatalyst and either UV or visible light at near-ambient temperature, both benzodioxole-derived and alpha-amino radicals form chiral centres with impressive yield and selectivity (figure 1).
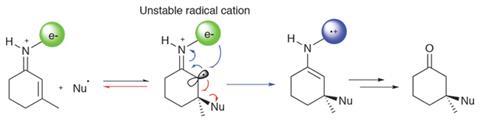
The team sought to exploit the early transition state for the formation of carbon–carbon bonds between nucleophilic radicals and electron-deficient alkenes. The ‘long’ forming bond in the transition structure is generally insensitive to steric hindrance. Conceptually, the two components of the reaction – forming nucleophilic radicals using a photocatalyst and activating enones with a chiral organocatalyst – are well established. But while the two reactive components are indeed complementary, the team identified that the initial addition product would be an unstable radical cation that is prone to reform the starting material, making a successful reaction highly unlikely (figure 2).
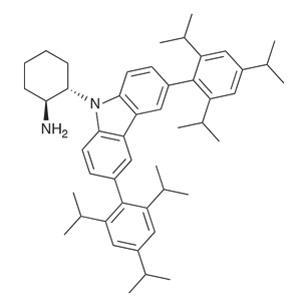
Taking inspiration from biological processes, the researchers decided to introduce a redox active component within the organocatalyst. This should provide a localised intramolecular electron pool to rapidly reduce the transient radical cation. This forms a neutral enamine species following tautomerisation, which ensures irreversible bond formation without erosion of stereochemistry. Carbazole heterocycle units provide an ideal electron pool, owing to their excellent electron donor capabilities and the high stability of carbazole radical cations – this explains why they find widespread use in LEDs and photovoltaic cells. Bulky substituents on the carbazole modify its reduction potential and provide increased enantiocontrol of the radical addition (figure 3).
Being the first effective catalytic strategy for forming asymmetric quaternary centres by radical conjugate addition, this work hopefully opens the door to a plethora of mild transformations that will provide access to unique quaternary carbon-based structures. However, the most groundbreaking aspect has to be the cleverly designed organocatalyst that tames the transient radical intermediate. This concept deserves to be seized upon, and will hopefully be developed in myriad ways to provide exquisite control in reactions where a loose electron has historically run riot.
Karl Collins leads a medicinal chemistry laboratory in Germany
References
J J Murphy et al, Nature, 2016, 532, 218 (DOI: 10.1038/nature17438)












No comments yet