Not all natural products are created equal. BRSM looks at a flexible route to some perennial favourites
Not all natural products are created equal. A cursory glance at the total synthesis literature from the past decade is enough to discover that some molecules – or families of molecules – attract a lot more attention than others.
Of course, some molecules are far more useful than others, and – at least in part – this is why targets like taxol, the bryostatins and the prostaglandins continue to enjoy the popularity that they do. However, it is clear that such ‘hot targets’ certainly span a spectrum of usefulness, and trying to explain why we choose to make the molecules we do is sometimes hard. I’m sure that many synthetic chemists look for the opportunity to do creative chemistry and overcome a challenge more than they look for the next blockbuster drug. However, this doesn’t mean just picking the biggest molecules; chemists usually prefer to choose targets that are just complex enough that an elegant or efficient synthesis is tantalisingly close to the current state of the art – or just out of reach.
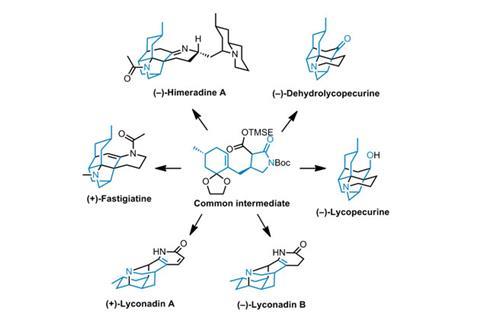
For this reason, many natural products enjoy only a brief period in the limelight, and are forgotten after the first synthesis or two. But a few remain in vogue even after decades of work and hundreds of publications. One such enduringly popular family is the Lycopodium alkaloids, some of which have been targeted by synthetic chemists for over 50 years. Indeed, this group of natural products – isolated from various species of club moss – has swelled to over 200 known members, and their syntheses continue to grace the pages of top journals.
Matt Shair and his group at Harvard University, US, have now set out enantioselective total syntheses of six members of this family, including four that had previously never been made in the lab. With their new, general strategy, they should be able to prepare an entire sub-family of the alkaloids built around a seven-membered ring. All the routes start from a key intermediate, which is quickly assembled in seven steps from the monoterpene pulegone and epichlorohydrin, which can then be converted into a large number of quite different targets (figure 1).
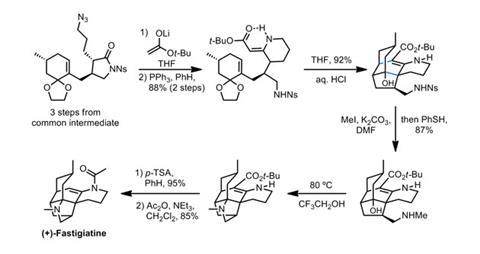
The first synthesis of fastigiatine begins with a few simple transformations of the common intermediate before the real chemistry begins. The team starts by opening the butyrolactone with the enolate of tert-butyl acetate to form a b-ketoester (figure 2). Reducing the nearby azide and condensing the resultant amide with the newly-formed ketone then forms a rather unusual-looking vinylogous carbamate, setting the stage for the key ring-forming step. Treating this intermediate with acid then cleaves the dioxolane protecting group, kicking off an enamine cascade that establishes the natural product’s entire carbon skeleton in a single step. From here, deprotecting and methylating the amine allows for a Mannich cyclisation to add the final pyrrolidine ring. Acidic decarboxylation removes the tert-butyl ester, and N-acetylation completes the synthesis.
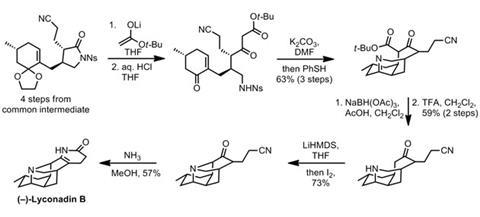
For lyconadin B, the group begins with a similar opening of a closely related butyrolactam, but follows this up immediately with dioxolane cleavage to reveal an enone (figure 3). Next, an interesting one-pot sequence consisting of a Michael addition, thiolate-mediated nosyl deprotection, and imine formation builds up two more rings with blinding speed. Reducing the imine and removing the unwanted tert-butyl ester then set the stage for completion. The final ring is forged by temporarily installing an iodide adjacent to the ketone, which is displaced by the amine to form the required C–N bond. Ammonolysis of the nitrile, and condensation onto the nearby ketone completes the natural product, and further heating in air transforms it into lyconadin A.
Check out the paper to see how the group synthesises another three members of the family.
BRSM (@BRSM_blog) is a postdoctoral researcher and blogger based in the US












No comments yet