New reactions need to belong in a synthesis, says BRSM, not be forced in for show
The relationship between new reaction development and total synthesis has always been an interesting one. Sometimes – although perhaps less frequently than is claimed – total synthesis begets new methods. But the flow of information is usually in the other direction. Indeed, even the most single-minded synthetic chemists are careful to keep abreast of new developments, so that they’re aware of all the options when planning synthetic routes. That said, sometimes the gap between a reaction being discovered and applied can span decades; even the venerable Diels–Alder reaction – undoubtedly one of the most powerful carbon–carbon bond forming reactions ever discovered – lay unused for 23 years before it was first used in a total synthesis.
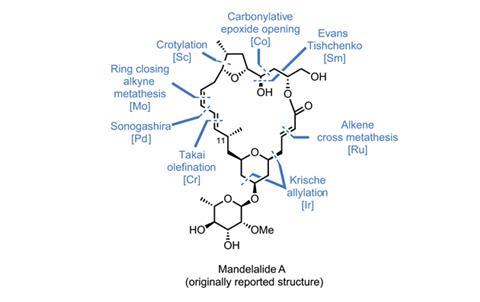
Of course, it’s not enough to merely use a new reaction; it needs to be appropriate, and not jarringly shoehorned into a synthetic sequence that has sacrificed aesthetics or efficiency to include it. The recent synthesis of the proposed structure of mandelalide A by Alois Fürstner’s group at the Max Planck Institute for Coal Research in Mülheim, Germany, is a good example of a well-designed route that seamlessly integrates some cutting-edge chemistry.1 The team synthesises the molecule in two halves, preparing the tetrahydrofuran (THF)-containing ‘northern’ domain and the ‘southern’ tetrahydropyran (THP)-containing domain separately, uniting them at the end of the route.
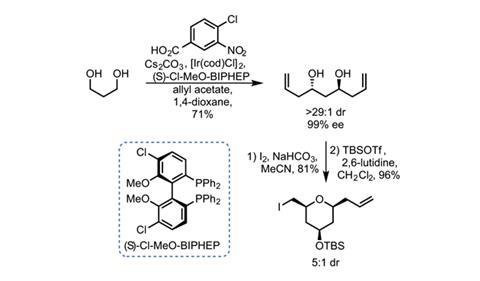
Constructing the southern portion hinges on some powerful chemistry developed by Michael Krische’s group at the University of Texas in Austin, US.2 This allows the team to quickly build up the required THP ring (figure 2). An off-the-shelf chiral iridium catalyst allylates both ends of 1,3-propanediol with essentially perfect diastereo- and enantiocontrol. This reaction is a very convenient way to allylate alcohols when the corresponding aldehydes are unstable. A good old fashioned iodoetherification gives the all-cis THP.
The northern domain is built up from (S)-glycidol, and its remaining stereocentres are set using an asymmetric crotylation, an Evans–Tishchenko reduction and a selenoetherification.
If you’ve read a Fürstner macrolide synthesis from the last decade or so, you might expect it to feature the group’s signature reaction: ring-closing alkyne metathesis. This highly chemoselective reaction is almost always performed on internal alkynes, as terminal alkynes are given to uncontrollable polymerisation and a host of other side reactions. The main drawback is that internal alkynes are harder to prepare than their terminal cousins, often requiring more steps and/or strongly basic conditions.
Although the first reports of internal alkyne metathesis appeared in the 1970s, the first catalyst that could reliably unite terminal alkynes wasn’t disclosed until 2012,3 and as a result remains largely untested. Before this work, no total synthesis had included metathesis featuring even a single terminal alkyne. But despite this, the Fürstner group still decided to use this reaction for the key macrocycle-forming step.
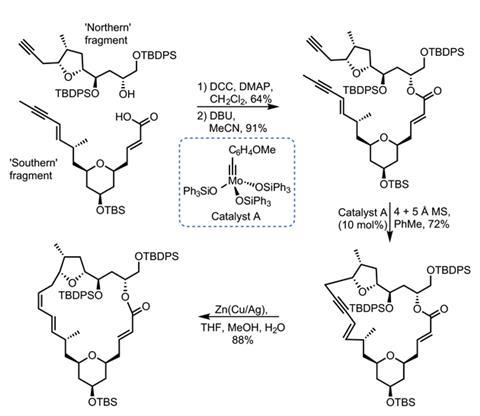
Joining together the two halves with an ester link proved surprisingly difficult. N,N’-dicyclohexylcarbodiimide (DCC) gives the best results, but strangely causes the adjacent olefin to move out of conjugation with the ester carbonyl (figure 3). A little base isomerises it back into the correct position. The stage is now set for the key metathesis step. Treating the diyne with a molybdenum catalyst produces the desired macrocyclic product in great yield. The resulting alkyne is then reduced to the Z-olefin, and just four more steps complete the target molecule.
Unfortunately, the spectroscopic data do not match that of the natural product, so further work is required to determine the true structure of mandelalide A. Still, with such an efficient, modular route in hand, it seems that the group is well set up to solve this problem.
BRSM (@BRSM_blog) is a postdoctoral researcher and blogger based in the US
References
1 J Willwacher and A Fürstner, Angew. Chem., Int. Ed., 2014, DOI: 10.1002/anie.201400605
2 Y Lu et al, Angew. Chem., Int. Ed., 2009, 48, 5018 (DOI: 10.1002/anie.200901648)
3 B Haberlag et al, Angew. Chem., Int. Ed., 2012, 51, 13019 (DOI: 10.1002/anie.201207772)












No comments yet