Paul Docherty ponders a tasty cheeseboard as a source of synthetic inspiration
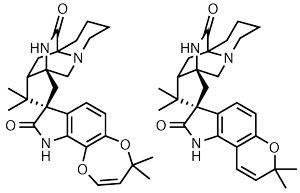
Natural product isolation is generally a tale of a journey to an obscure or inaccessible location, followed by pulping a harmless plant or marine sponge to get at compounds made by some bacteria hiding out in the core. Isolating the marcfortines was probably somewhat more mundane, however, potentially involving a trip to the local cheesemonger and some detailed analysis of the blue cheese section. Found as secondary metabolites in the Penicillium roqueforti fungus that gives Roquefort its blue veins, their complex heptacyclic ring systems give organic chemists something to ponder over while enjoying a tasty cheeseboard.1 Derived from the amino acid tryptophan, a monoterpene section and a cyclic amino acid moiety, the central quaternery spiro-ring junction represents a considerable synthetic challenge – one that Barry Trost of Stanford University, US, thought particularly suited to his group’s trimethylenemethane cycloaddition chemistry.2
Originally developed by Trost in the late 1970s, the palladium catalysed cycloaddition of trimethylenemethane (TMM) is a powerful method for constructing ring systems. Since then, several improvements and modifications have appeared – most importantly an asymmetric variant.3 And it’s this significant modification which makes TMM cycloaddition so suited to this synthesis. However, TMM itself is both rather unusual and vanishingly reactive, so is generally produced in situ. Thankfully, three decades of practice have ensured that Trost and his team are experts in manipulating this volatile intermediate.
First of all, the team completed a neat and concise racemic synthesis of marcfortine B, using the TMM cycloaddition as a critical step. Working from a known oxindole, a dehydrating aldol reaction installs the exocyclic alkene with its pair of matching methyl groups, and a quick Boc protection subdues the reactive nitrogen atom. Applying the group’s standard TMM cycloaddition reaction conditions with an appropriate source of the reactive intermediate generates the desired cyclopentane and creates the critical 5,5-spiro ring junction in exceptional yield. The team used this success as a platform for completing marcfortine B in a reasonably impressive 25 steps.
Barely stopping for breath, the team restarts the ring-building masterclass, moving on to marcfortine C.
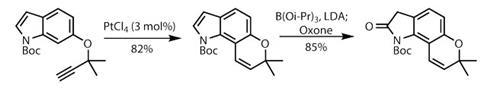
The lab work begins with the relatively simple aromatic region, starting with a modified indole and elaborating with a propargylation reaction (figure 1). A mild, metal-catalysed Claisen rearrangement closes the dihydropyran ring, before the indole is oxidised to a lactam in two steps, firstly forming a boronate and then oxidising with the mild Oxone reagent.
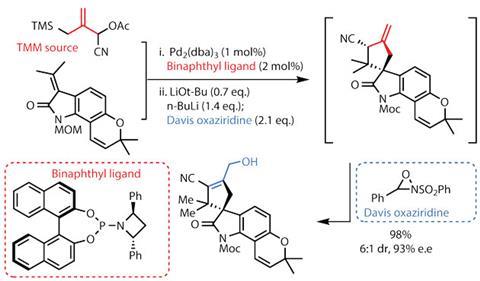
The team then moves back to more familiar territory, this time using a modified TMM-cycloaddition where the TMM source bears an additional nitrile group (figure 2), adding to the complexity of an already intricate transformation. A productive and mercifully brief ligand screening exercise highlighted a somewhat unusual binaphthyl phosphoramidite as the best partner for the palladium catalyst, giving a remarkable yield and good stereoselectivity in the 5,5-spiro ring system. Oxidising the deprotonated allyl cyanide with an oxaziridine reagent developed by Franklin Davis of Temple University, US, creates a neat allylic alcohol synthetic handle.
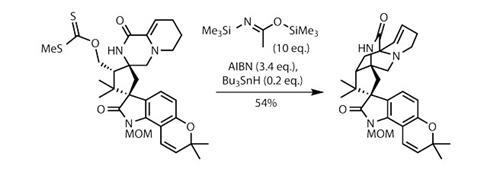
This alcohol is put to work immediately as its mesylate derivative, which is displaced by an amidopiperidine ring. Manipulating this region of the molecule took several steps and some careful optimisation, eventually closing the piperazin-2-one ring, and converting the nitrile group into a xanthate ester. This slightly unusual sulfur-based functionality is a bit of a one trick pony, but it does that trick rather well. Under (interestingly modified) radical-inducing conditions (figure 3), a somewhat risky but impressive late-stage cyclisation reaction forms the final ring in impressive yield, leaving the team only an olefin reduction and deprotection away from marcfortine C.
Remarkably, the team completes this breakneck asymmetric synthesis in 19 steps – even fewer than the racemic version. But what really stands out is their mastery of a rather unusual reaction.












No comments yet