Late stage dimerisation is a tantalisingly elegant but risky strategy for total synthesis, says BRSM
It’s a somewhat surprising assertion that almost a fifth of natural products are thought to include a dimerisation step somewhere in their biosynthesis.1 Of course, this is not always apparent at first glance – it’s often masked by subsequent modifications – and the ability to spot such ‘hidden symmetry’ can often lead to strikingly effective total syntheses. Indeed, Aristotle once famously observed that ‘the chief forms of beauty are order and symmetry’, and given the recent completion of two large, dimeric natural products,2 it seems that the synthetic community is inclined to agree.
When tackling such targets, most chemists will take the somewhat obvious, convergent approach of making a monomer and then dimerising it towards the end of the route. This approach – which often mimics the natural route – is efficient, direct, and appealing in its simplicity. The problem is that the reality is rarely as simple as it looks on paper. The biosynthetic conditions are often difficult to reproduce in a round-bottomed flask, especially if enzymes are involved. Despite the aesthetic appeal of copying nature, yields are typically low.
Furthermore, large targets often have monomers that take many steps to prepare, forcing the synthetic chemist to gamble months or years on a risky key step, and nebulous theoretical predictions, with few alternatives in the event of failure. One powerful reminder of the commitment involved in this kind of strategy comes from Seth Herzon at Yale University, US. His team tried over 1500 iterations of the key dimerisation in a biosynthetically inspired route to lomaiviticin aglycon, all for a final yield of just 26%.3
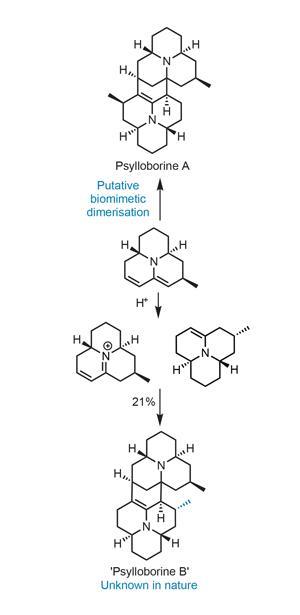
Another example of a late stage dimerisation going awry can be found in the synthesis of psylloborine A from Scott Snyder and coworkers, now located at the Scripps Research Institute in Florida. When they attempt the key step, the dimer they obtain is not the desired target, but a previously unknown molecule where the monomers come together with the wrong regiochemistry (figure 1). Unperturbed, the team christened the new molecule psylloborine B (in case it should ever be found in nature) and started to think about a new way to approach psylloborine A.
Their new plan – dubbed ‘intramolecular dimerisation’ – is a new twist on the time-honoured tactic of tethering molecules together to enable reactions where the intermolecular version would be impossible. In practice, this involves preparing a protected diketone substrate that is set up to quickly generate the linked enamine and iminium species required for the pseudo-dimerisation.
The group quickly assembles the molecule using fairly standard chemistry. Then, through an ingenious pair of cascade reactions, converts it to the natural product in just three steps, forming five bonds, five rings and four stereocentres in the blink of an eye (figure 2).
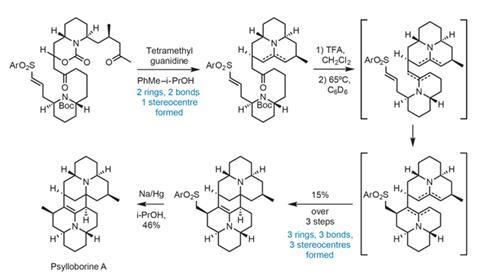
First, treating with base simultaneously generates an enone and deprotects the top amine. This then condenses with the nearby methyl ketone – forming one piperidine ring. The resulting enamine adds to the new enone, forging the second ring, all in one pot. Deprotecting the bottom amine and heating the mixture then begins a second, similar cascade to complete the remaining rings. First an enamine is formed by the amine condensing onto the remaining ketone. This cyclises onto the nearby vinyl sulfone, and the final carbon–carbon bond forms to complete the polycyclic skeleton. With the synthesis all but over, all that remains is removal of the sulfone group that enabled the second cascade. Congratulations to the team on a spectacular endgame!
BRSM (@BRSM_blog) is a postdoctoral researcher and blogger based in the US












No comments yet