Sometimes it’s worth building aromatics instead of buying them, says BRSM
It’s likely that organic chemists have been practising retrosynthesis in one form or another for at least a century, and certainly for decades before E J Corey formalised the concept in the mid-1990s. And while it’s hard to know now exactly how the chemists of the early 20th century planned their attacks on nature’s molecules in the days before synthons and our appropriation of the logicians’ open-ended arrows, today’s chemists still benefit hugely from their contributions to the chemical literature.
Nowadays, many common functional groups have well-established disconnections attached to them – some of which would seem near-magical to chemists of 50 years ago. That said, there remain plenty of bad disconnections that are best left unmade. Some moieties are still best bought in as commercial building blocks, or included through judicious choice of starting materials.
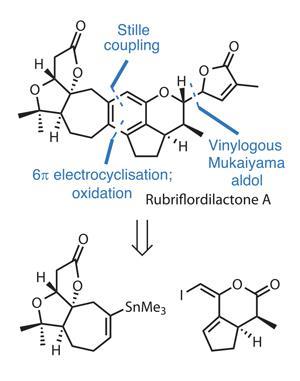
Aromatic hydrocarbons usually fall firmly into this latter class of ‘buy, don’t make’ functional groups, particularly if there’s only one in the compound of interest. Of course, there are exceptions; when a very highly substituted ring system is required, or a single aromatic ring is located in the very centre of a target molecule, chemists may start to nervously size up constructing it from scratch. One example where this somewhat uncommon tactic fits perfectly is in the synthesis of rubriflordilactone A by Ang Li and coworkers at the Shanghai Institute of Organic Chemistry in China.
The team has devised a creative retrosynthesis that cuts the target in half by disconnecting the central benzene ring back to a triene. The projected late-stage combination of a Stille coupling, electrocylisation and oxidation gives two equally sized advanced fragments to work with – and sidesteps a lot of tedious classical aromatic substitution chemistry along the way (figure 1).
First targeting the vinyl iodide fragment, the group starts from 2-iodocyclopentenone (figure 2), using a Corey–Bakshi–Shibata (CBS) catalytic reduction to introduce asymmetry in this half of the molecule. A Sonogashira coupling introduces an alkyne, and a Johnson–Claisen rearrangement relays the chiral information from the alcohol stereocentre across the ring and creates two new stereocentres. Iodolactonisation of the deprotected alkyne and carboxylic acid with N-iodosuccinamide gives the desired fragment in five steps.
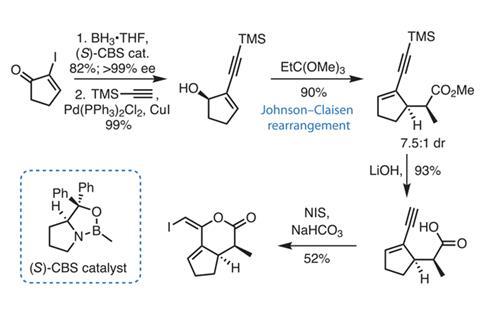
After a slightly longer preparation of the stannane fragment – using a catalytic Diels–Alder followed by ring expansion to make the tricky 7-membered ring – the team turns its attention to unifying the two. First, a Stille coupling brings the halves together to form a triene in excellent yield. Heating this unstable intermediate under oxygen (figure 3) accomplishes both electrocyclisation and oxidation of the resulting diene in a single step.
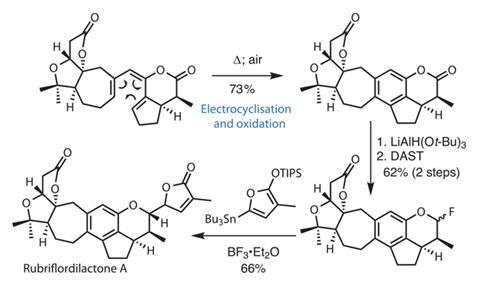
Lithium trimethoxyaluminium hydride selectively reduces the six-membered ring lactone, leaving the five-membered lactone untouched. The group had planned to couple this lactol with a silyloxyfuran in a vinylogous Mukaiyama reaction to attach the final butenolide ring. However, these attempts gave only ring-opened products, presumably owing to the strain in the fused polycyclic system and the stability of the phenol as a leaving group.
Converting the lactol to the corresponding fluoride with diethylaminosulfur trifluoride (DAST), gives a new electrophile that is (unfortunately) entirely unreactive to a number of silyloxyfurans and Lewis acids. Fortunately, pepping up the nucleophilicity of the furan component by attaching a tributylsilyl group allows the reaction to proceed, delivering the natural product in good yield. Well done to the group on making this unusual disconnection work so well!
BRSM (@BRSM_blog) is a postdoctoral researcher and blogger based in the US
References
J Li et al, J. Am. Chem. Soc., 2014, 136, 16477 (DOI: 10.1021/ja5092563)












No comments yet