There’s more to bonding than covalent, ionic and the lines we draw between atoms on paper. Philip Ball takes on the expanding list of chemical connections
Almost 100 years ago, the American chemist Gilbert Lewis proposed that chemical bonds arise from the sharing of pairs of electrons between atoms. In a 1916 paper,1 he portrayed atoms as cubes with electrons at their corners, and argued that they accumulate an electron at every corner by sharing edges with other atoms. It was a dramatic shift from the prevailing notion of chemical bonding as an electrostatic interaction caused by the transfer of electrons between atoms. Not everyone liked the idea, but over the ensuing two decades Linus Pauling showed how electron sharing could be described by the new theory of quantum mechanics, making Lewis’ picture central to modern chemical bonding theory.
It still is. But since that time, variants on Lewis’ theme have proliferated. Dative bonds (now more generally called donor–acceptor bonds), where both electrons in the pair come from the same atom, and hydrogen bonds, where the interaction of a hydrogen atom with a lone pair is primarily electrostatic, are standard undergraduate fare. However, thanks partly to improved methods of determining molecular structure and partly to greater sophistication of quantum-chemical theory, new modes of bonding are still being discovered, and controversies continue to rage over whether or not a particular pair of atoms can be considered united by a bond. So is Lewis’ concept of a bond still fit for purpose in the 21st century?
Shifting bonds
Traditionally, chemical bonds have been classified as either Lewis’ electron-sharing variety (covalent) or the older electron-swapping sort (ionic). But not only have these distinctions sometimes become blurred, other new bonding types are being identified. One such is the charge-shift bond.2 Here the electrons are not simply shared or transferred, but undergo strong fluctuations that give rise to a bonding interaction redolent of old ideas about resonance structures in Pauling’s valence-bond theory.
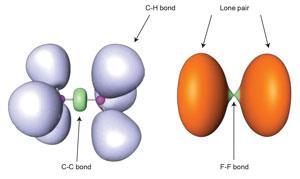
Charge-shift bonding is not some quirk of rare and exotic molecules, but is present even in such routine unions as the fluorine molecule, F2. This is typically described in high-school chemistry as a simple example of covalent electron-sharing, but the purely covalent contribution to the bond turns out to be repulsive. The atoms can instead be considered to be held together by resonant mixing between ionic and covalent forms, as Sason Shaik, David Danovich, Philippe Hiberty and their coworkers showed in 2005.3
One way to think of this process is that, as the covalent bond pulls the atoms together, their shrinkage causes a rise in electron kinetic energy sufficient to offset the potential-energy stabilisation of bond formation; the ionic·covalent resonance energy restores the balance. Other descriptions of chemical bonding, while using a different formal framework for thinking about bonds, also identify charge-shift bonds as something distinct from traditional covalency. As a result, Shaik, Hiberty and colleagues argue that charge-shift bonding ‘constitutes a large and distinct class of bonding alongside the two classical families’.2
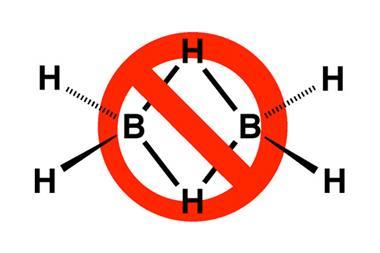
Even apparently familiar bonds still spring surprises. The donor–acceptor bond, in which one atom donates an electron pair to another, is common in the bonding of ligands to transition metals, but somewhat rare among main-group elements. One example of the latter is the compound carbodiphosphorane, C(PPh3)2. When this was first made in 1961, it was assumed that the phosphorus atoms were bound to the central carbon via double bonds. But in 1978, the molecule was found to be bent rather than linear.
It wasn’t until 2008 that the reason was fully understood, when quantum-chemical calculations by Gernot Frenking and Ralf Tonner of the University of Marburg in Germany showed that in fact it should be considered a donor–acceptor complex in which the phosphorus atoms donate lone pairs to a neutral carbon atom in a formally zero-valent excited singlet state – that is, with its valence-shell electrons already gathered into lone pairs.4 Other compounds of this general type CL2 have now come to light, and Frenking and Tonner propose calling them carbones.5 Analogues with silicon and germanium in place of carbon have also been identified. Frenking stresses that such compounds ‘can easily be fitted in the traditional category of donor·acceptor bonding’ – they just present a rather odd and unforeseen example of it. Such bonds can in fact also be regarded as another variant of charge-shift bonds.
One such carbone, C(BH)2, turns out to be particularly odd, since it may exist in two isomeric forms in which each atom is joined to the same partner but which differ in their mode of bonding: in one isomer the boron atoms are linked to carbon by double bonds and the molecule is linear, while the other is a carbone with donor-acceptor bonds, and is bent at an almost perfect right angle.6
Filling a hole
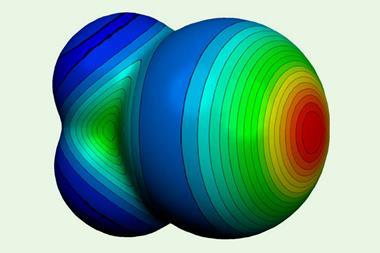
Other familiar bonding types are getting a makeover too. A recent recommendation by the International Union of Pure and Applied Chemists for a redefinition of the hydrogen bond took into account reconsiderations of significant covalent contributions to what has traditionally been regarded as a basically electrostatic interaction. And the ‘halogen bond’, long considered a non-covalent interaction in some ways analogous to the hydrogen bond, also has a new image. These bonds, which weakly link a halogen on one molecular group to an electronegative atom on another, have been known since the 19th century, although the terminology of halogen bonding came into vogue only in the 1970s. But now they have been recast as a subset of a more general class called s-hole bonds, in which the negative partner (typically a lone pair on a Lewis base, such as nitrogen or oxygen) interacts with a region of positive charge on the halogen7– making them somewhat analogous to donor–acceptor complexes.
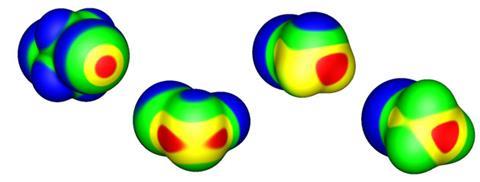
This positive dimple or s-hole on the halogen, at the opposite pole to its covalent bond, was first identified by Peter Politzer and Jane Murray of the University of New Orleans and their coworkers.7,8 It seems a peculiar property for a type of atom generally considered electronegative, but is a straightforward consequence of the fact that the electron density accumulates in the vicinity of the covalent bond – especially for more polarisable halogens such as bromine and iodine. This redistribution of charge is accompanied by an ‘equatorial’ band of negative charge between these poles, which means that two halogens can even interact electrostatically like with like, the positive part of one aligning with the negative region of the other. The interaction is augmented by dispersion (van der Waals) forces that arise from correlations between fluctuations in the electron densities on the two interacting atoms.
Non-covalent interactions with negative sites (including same-atom pairings) have also been observed for group 15 and 16 elements, and have become called pnictogen and chalcogen bonds respectively. Examples of such bonding were in fact spotted up to four decades ago, but only recently have they been afforded much attention. As Politzer, Murray and colleagues have shown, they too can be classed as s-hole bonds.
They believe that the s-hole concept ‘unifies our understanding of a lot of seemingly different interactions, involving different groups of the periodic table and different negative sites’ – which includes hydrogen bonding. ‘There is a tendency to want to view all of these as separate and different interactions,’ they say – but add that ‘this is artificial and misleading’.

The array of new classes or variants of bonds seems sometimes to grow by the week. There are, for example, f-bonds of f-block elements,9 parallel-spin bonds of metal atoms,10 and Möbius bonds of twisted aromatic rings.11 Such developments have stimulated some researchers to foresee striking changes in the way chemists think about assembling atoms and molecules, using linkages other than the purely covalent. The essential role of weak interactions in receptor·ligand binding in molecular biology gives ample reason to consider any addition to the chemists’ repertoire of ‘loose grips’ a potentially useful one. Indeed, halogen bonds have been used already to enable molecules to self-assemble into regular arrays for crystal engineering, with potential applications in optics and electronics.12 Both they and chalcogen (sulfur–oxygen) bonds have also been identified in biological systems, which suggests that they might be used in drug design.13
To bond or not to bond?
One of the key ambiguities in chemical bonding lies with the relationship between the strength and length of a bond. Chemists are accustomed to the idea that strong bonds tend to be short, and weak ones long. But this doesn’t always seem to mean that atoms that are close enough together to be bonded necessarily are. By the same token, some bonds can seemingly be stretched beyond their normal breaking point without ‘snapping’.
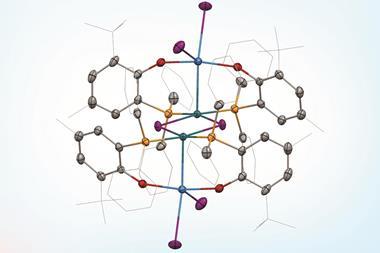
Take the case of the cluster Cu3 S2, which appears in a class of organometallic compounds first explored by William Tolman of the University of Minnesota in the US. Here each of the two sulfur atoms is joined to each copper atom in a flattened trigonal bipyramid. The question is whether the sulfur atoms are also bonded to one another down the central axis of the cluster. Calculations by Roald Hoffmann at Cornell University in the US, Carlo Mealli at the Institute of Organometallic Chemistry in Florence, Italy, and their coworkers seemed to suggest there was,14 but Santiago Alvarez at the University of Barcelona in Spain believed there wasn’t. After debating their differences at length,15 the three researchers concluded that ‘Perhaps the lesson is not that people disagree, but that the concept of a chemical bond is not so simple’ – not least because its presence or absence can be evaluated by several different experimental and theoretical criteria, which are not necessarily consistent.
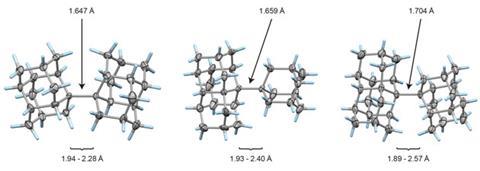
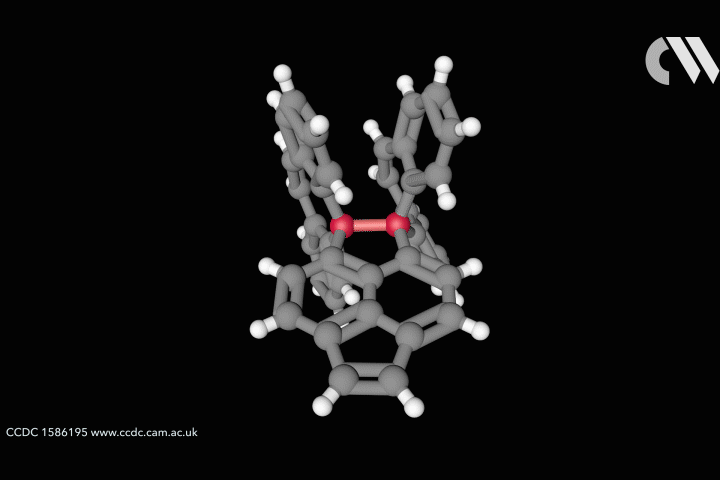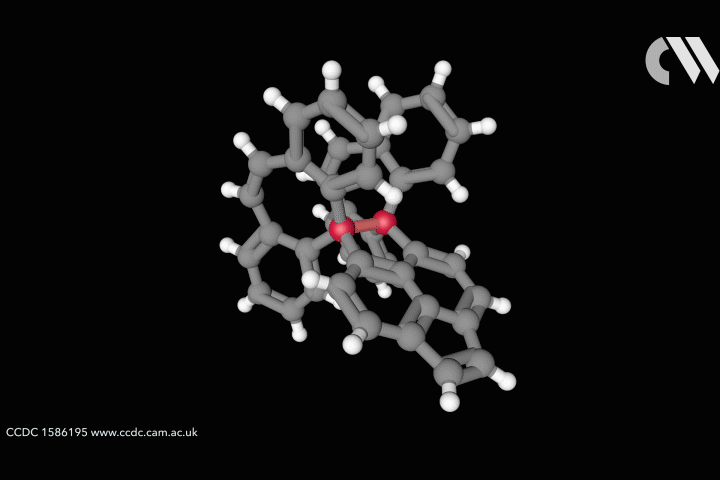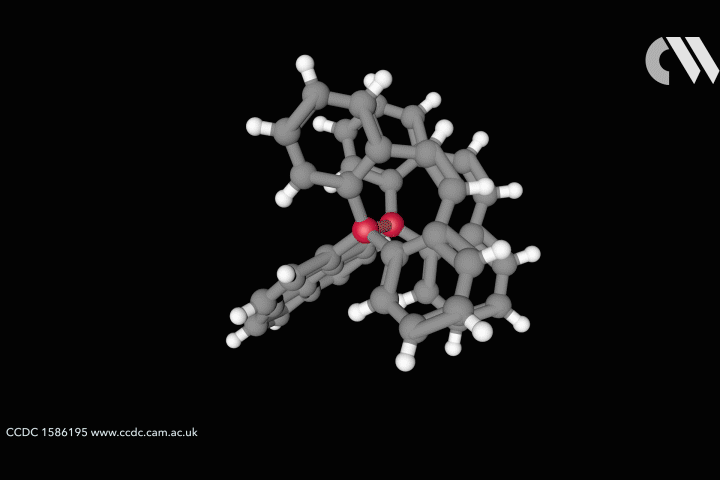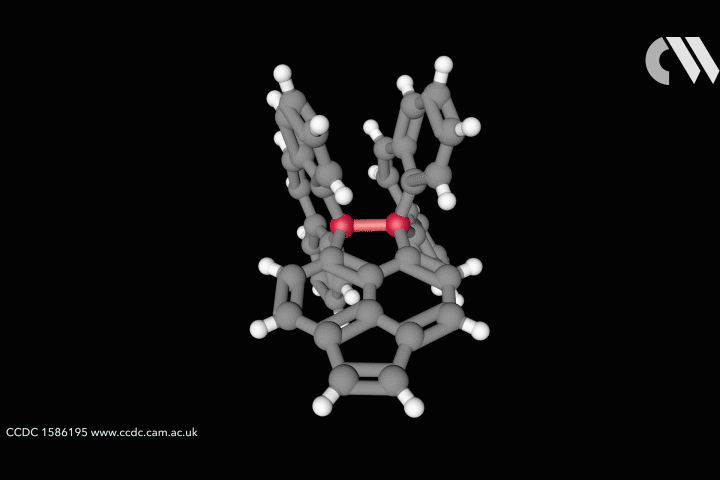
The whole issue of regarding a bond as a link between two specific atoms is complicated by the recent work of Peter Schreiner of the University of Giessen, Germany, and his coworkers. They found that unusually long carbon–carbon single bonds can be sustained in some exotic molecules, in which they connect very bulky fragments of the diamond network called diamondoids.16,17 While the carbon–carbon single bond in hydrocarbons is typically about 1.54Å long and rarely deviates from that by more than 10%, in Schreiner’s compounds it could be up to 1.704Å – the longest ever observed. Nonetheless, they are quite stable: the compounds don’t decompose until temperatures of above 200°C. This is in spite of the fact that carbon·carbon bonds of 1.65Å in some sterically crowded compounds have previously been found to be very weak.
The reason, the researchers say, is because the diamondoid substituents sit opposite one another across the bond with a lot of surface area juxtaposed, so that dispersion interactions can help bind them together. In effect, these surfaces are ‘weakly glued’, so that the long covalent bond between them remains viable. In fact, quantum-chemical calculations by Schreiner’s team suggest that, if the juxtaposed surfaces are large enough, a single connecting carbon·carbon bond can still contribute a well-defined binding energy even at the almost surreal length of over 4Å.
‘Our common bonding models, both theoretically and experimentally, almost exclusively build on the analysis of relatively small molecules, where the dispersion interactions play a rather minor role,’ says Schreiner. But his results and other quantum-chemical computational schemes ‘show that dispersion cannot be neglected for increasingly larger molecules’, he says.
Defining difficulties
This raises the question of whether the carbon–carbon bond in these compounds really is a bond at all in the traditional sense. It’s almost like suggesting that, because there’s a thin thread stretching tautly from one building to the adjacent one, it must be binding them together. At the very least, Schreiner’s results challenge the traditional tendency to regard a bond as an isolated entity, somehow transferrable from one molecule to another. ‘We’ve believed that isolated-bond picture because our bonding models originally all go back to diatomic systems that we take as a reference,’ says Schreiner. Even a picture that accommodates delocalised and nonclassical bonds has tended to attribute the links between specific atoms to a small specific subset of electrons. But Schreiner says his findings show that a bond ‘can no longer be considered a transferrable property from one molecule to another but rather depends on the nature of the entire molecule’.
Calling any attractive interaction a bond threatens to take us back to notions of ‘chemical affinity’
How to define a chemical bond in the first place has long been a point of contention, and some of the quirky new bond types are now forcing that issue. Some researchers, notably the late Richard Bader at McMaster University in Hamilton, Canada, have argued that any attractive interaction between two atoms should be considered a bond – even the van der Waals or dispersion forces that exist between all atoms, so long as they can hold the atoms concerned into stable unions. Bader’s ‘quantum theory of atoms in molecules’ (QTAIM) purports to offer a unified picture of all chemical bonding in terms of the topology of electron density in molecules, which both identifies and classifies each bond. Others find this picture of limited value. Calling any attractive interaction a bond, says Shaik, threatens to take us back to 18th-century notions of ‘chemical affinity’.
Yet Alvarez feels that it’s impossible to be dogmatic about the bonding status of van der Waals interactions. His recent analysis of the ‘van der Waals territories’18– the typical distances that separate atoms united just by these interactions – led him to conclude that ‘depending on the pair of chemical elements chosen, and sometimes on their oxidation state or other variables, there may sometimes be a continuum of interatomic distances that does not allow us to establish a frontier between bonded and non-bonded atoms’.
Lewis’ legacy

Does all this mean that we should rethink the concept of a bond? The approaching centenary of Lewis’ seminal paper is sure to provoke discussion about that. Some researchers advise caution about too much revisionism. ‘I think there is a lot of hype recently about new bonds like halogen bonds, pnictogen bonds, chalcogen bonds, beryllium bonds, and whatnot,’ says Shaik. ‘We have to be careful not to get trapped in this fashion.’ He feels that it is perfectly clear and reasonable to describe some weak attractions between atoms simply as ‘intermolecular interactions’, rather than giving them the status of chemical bonds. ‘The way I look at bonding is simple,’ he says. ‘There are interactions which pair up electrons, and give rise to the entities we call molecules, which are glued by these chemical bonds. Then there are interactions between these molecules that are responsible for the formation of mesoscopic and macroscopic matter. These interactions are usually weak and they usually do not involve electronic reorganisation such as electron pairing. To call these a “Z-bond” adds no physical or chemical insight other than being a matter of taxonomy.
‘I think that quantum chemistry has so far supported very much the Lewis model of electron pairing,’ he asserts. ‘True, the picture has been broadened and there are exceptions, but most molecules are bonding as Lewis said. I think the electron-pairing mechanism will survive as a major mechanism of bonding.’
Others are more ambivalent. ‘At present I don’t see a clear way to make a new definition of the chemical bond that is at the same time practical and all-encompassing,’ says Alvarez. ’Probably we need to develop new ways of looking at interatomic interactions, or maybe the improvement of visualisation techniques at the atomic scale will provide relevant information to help us develop a new systematic scheme.’ But he agrees that the Lewis model still ‘works well enough for the largest part of the “real” chemistry, and is a good starting point for the analysis of anomalous cases’. In other words, ‘classifying bonds into “covalent” and “non-covalent” is still something we can teach in high school and early courses at college, since this provides a useful first approximation’. Just don’t believe everything you learn there.
Philip Ball is a science writer based in London, UK
References
1 G N Lewis, J. Am. Chem. Soc., 1916, 38, 762 (DOI: 10.1021/ja02261a002)
2 S Shaik et al, Nat. Chem., 2009, 1, 443 (DOI: 10.1038/nchem.327)
3 S Shaik et al, Chem. Eur. J., 2005, 11, 6358 (DOI: 10.1002/chem.200500265)
4 R Tonner and G Frenking, Chem. Eur. J., 2008, 14, 3260 (DOI: 10.1002/chem.200701390)
5 G Frenking and R Tonner, Pure Appl. Chem., 2009, 81, 597 (DOI: 10.1351/pac-con-08-11-03)
6 S R Barua et al, Chem. Eur. J. in press.
7 T Brinck, J S Murray and P Politzer, Int. J. Quantum Chem., 1992, 44, 57 (DOI: 10.1002/qua.560440709)
8 P Politzer, J S Murray and T Clark, Phys. Chem. Chem. Phys., 2013, 15, 11178 (DOI: 10.1039/c3cp00054k)
9 S G Minasian et al, Chem. Sci., 2014, 5, 351 (DOI: 10.1039/c3sc52030g)
10 D Danovich and S Shaik, Acc. Chem. Res., 2013, DOI: 10.1021/ar4001422
11 C S M Allan and H S Rzepa, J. Org. Chem., 2008, 73, 6615 (DOI: 10.1021/jo801022b)
12 V Amico et al, J. Am. Chem. Soc., 1998, 120, 8261 (DOI: 10.1021/ja9810686)
13 P Auffinger et al, Proc. Natl. Acad. Sci. USA, 2004, 101, 16789 (DOI: 10.1073/pnas.0407607101)
14 C Mealli et al, Angew. Chem. Int. Ed., 2008, 47, 2864 (DOI: 10.1002/anie.200705296)
15 S Alvarez, R Hoffmann and C Mealli, Chem. Eur. J., 2009, 15, 8358 (DOI: 10.1002/chem.200900239)
16 P R Schreiner et al, Nature, 2011, 477, 308 (DOI: 10.1038/nature10367)
17 A A Fokin et al, J. Am. Chem. Soc., 2012, 134, 13641 (DOI: 10.1021/ja302258q)
18 S Alvarez, Dalton Trans., 2013, 42, 8617 (DOI: 10.1039/c3dt50599e)








No comments yet