
Andy Extance finds out how chemists are studying processes lasting trillionths of a second – and even less – using laser-based pump–probe experiments
Clad in green-tinted safety goggles, Susan Quinn and Mike Towrie are fine-tuning almost incomprehensibly brief bursts from the LIFEtime laser system housed in the black cabinets snaking around them. In the open cabinet in front of Quinn, an optical cell containing orange crystals circles rapidly, like a tiny, single-chair Ferris wheel on fast-forward. Using a screwdriver, Quinn is twisting the cell’s position to get the crystals properly aligned in the laser beam. Towrie peers up at a screen showing wriggling lines of infra-red absorption spectra, clicking buttons to change when laser pulses hit the crystals.
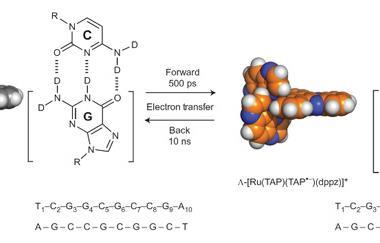
Quinn, from University College Dublin in Ireland, wants to understand more about the very first steps of DNA damage caused by light. That knowledge is valuable because it should help better understand how solar ultraviolet (UV) radiation can mutate human DNA and cause cancer, for example. She’s visiting the Rutherford Appleton Laboratory (RAL) in Didcot, UK, where Towrie leads the Central Laser Facility group studying ultrafast chemical processes. Towrie is helping Quinn and her colleagues investigate DNA crystals, whose orange colour originates from the ruthenium complexes bound to them. For Quinn, the lines on the screen are a scientific ‘wow’ moment – they represent snapshots lasting picoseconds, just trillionths of a second (10–12s).
Pairs of laser blasts hold the key to the scientific superpower the scientists use to glimpse events far beyond human ability to perceive unaided. The first blast, known as the pump, excites a molecule or triggers a reaction and the second, known as the probe, collects information after a tiny time lapse. At RAL and in hundreds of other laboratories across the world, pump–probe spectroscopy uses various wavelengths to collect in-depth information on the fastest chemical processes. Yet as the approach pushes the limits of what it’s possible to know, it enters new realms of practical challenges.
Pump–probe approaches build on pre-existing spectroscopy expertise, explains Marc Vrakking, director at the Max Born Institute for Nonlinear Optics and Short Pulse Spectroscopy in Berlin, Germany. Yet they address questions very differently. ‘In the past, most research prepared molecules prior to a chemical reaction, then looked in great detail afterwards at the products,’ Vrakking says. ‘That’s a bit like seeing a car wreck: what you see on the pavement doesn’t necessarily tell you how this accident really took place.’ Most pump–probe experiments are more like exactly replicating that car wreck over and over, taking a snapshot at a different stage of the process each time. Compiling those snapshots effectively creates an animated movie.
Vrakking changed lanes from conventional spectroscopy to pump–probe experiments in 1994, a manoeuvre enabled by advances in laser technology made shortly before. Lasers producing pulses lasting femtoseconds (fs) – a femtosecond being 10–15s – were still very expensive and difficult to use, and therefore relatively few groups were involved, he says. But today, there are user-friendly titanium-doped sapphire lasers that emit near-infrared (NIR) 680–1080nm femtosecond light pulses and use optical parametric amplifiers (OPAs) to boost their power and alter the wavelengths produced. ‘They’re not cheap but they have become affordable to many,’ Vrakking adds.
Coming up TRMPS
RAL’s LIFEtime system pairs femtosecond lasers and OPAs for time-resolved multiple probe spectroscopy (TRMPS). As little as 300fs after the pump blast, TRMPS begins a continuing series of probes, directly recording successive spectra, like individual frames of a movie. That continuity is especially useful, explains Quinn’s colleague John Kelly from Trinity College Dublin, because in the past recording data on different timescales from milliseconds down to femtoseconds needed separate instruments. Not only did that mean multiple experiments had to be run independently, they were often done in multiple countries, bringing logistical challenges. ‘Bolting it all together was tough, but LIFEtime provides it all in one seamless piece,’ Kelly explains.
The screen in front of Towrie is showing an average of the 100,000 IR spectra taken in a second. The UV pump blast’s energy is absorbed by the DNA crystal, putting it into a temporary excited state, whose lifetime can be monitored by IR probes. Towrie changes the delay between the pump and the first probe from 2ps to 100ps, and the signal from the excited state gets progressively smaller and disappears.
While detailed analysis will happen later, looking directly at spectra helps avoid problems like ‘overcooking the samples’, Towrie explains. To produce strong enough signals, pump–probe experiments must use high laser powers that risk destroying the material being investigated. ‘A major part of my job is to design a world-leading instrument that gives as much information out of this little valuable sample without bringing a tear to Susan’s eye,’ Towrie says. The Lego-like ferris wheel contraption is a surprisingly low-tech solution to this problem, rotating to expose different crystals to the laser. ‘To stop the sample frying it’s important that when pulses come in they’re not hitting the same part of it repeatedly,’ Quinn explains. On timescales shorter than microseconds crystals move a negligible distance between the pump and probe, ensuring the excited molecules are the ones that are measured, before they move on.
Delivering femtosecond-level synchronisation between laser pulses involves precision electronics, but the final level of control Quinn and Towrie exploit is again relatively simple. A grey aluminium plate with a mirror mounted on it is attached to a folded-over caterpillar track. Towrie changes the probe delay by a nanosecond (ns) – that is, a billionth of a second. The caterpillar track lays itself out flat, sliding the plate and mirror along to the opposite end of the cabinet they’re in. Because light travels 30cm in a nanosecond, changing the distance between laser source and sample offers easy timing control. ‘To get femtosecond accuracy you have to move a few tenths of a micron,’ Towrie adds. ‘We can get down to 1fs steps quite easily.’

Seeing that the DNA excited state disappears in picoseconds on Towrie’s screen is notable, stresses Kelly. That’s because excited states of organic molecules typically last much longer, around a nanosecond. And thanks to Christine Cardin and James Hall from the University of Reading, UK, they’ve been able to combine such insights with x-ray crystallography data. Collected at the Diamond synchrotron in the building next door to LIFEtime, the crystal structures have pinpointed where the excited state forms. An electron transfers to the ruthenium complex from DNA at a specific guanine base residue, a first step that could continue on to an oxidation reaction.1 ‘Guanine oxidation leads to a change in the nature of the base – mutations, essentially,’ Kelly says. In human cells, similar mutations could be a first step towards cancer.
Fourier parks the bus
While the researchers are seeking as much information as they can get about processes like this, a trade-off comes into play when looking at shorter timescales, Kelly says. The duration of a chemical process and its absorption’s ‘bandwidth’ – the size of the slice of the electromagnetic spectrum it encompasses – are linked by a Fourier transformation. That means the faster the processes, the broader the absorptions, and the harder it is to pick out individual features in the spectra. ‘When you get down to a few femtoseconds you’re in real trouble,’ Kelly says. According to Quinn, this ‘parks the bus’, creating a big obstacle for getting chemical information.
However, another manifestation of this effect is also essential for the RAL’s infrared pump–probe instruments, because short pulses span a range of laser light wavelengths. ‘The lasers are inherently a stable broadband infrared light source for spectroscopy,’ Towrie explains. ‘That’s one of their key advantages.’
The effect allows scientists a luxurious observation window that can capture features from multiple parts of the structure being analysed, Quinn explains. ‘You can actually see if the energy is going from one part of the molecule to another,’ she enthuses. ‘That is really useful if you’re trying to understand how molecules respond to light energy at the most fundamental level. It helps with loads of different applications.’ The laser systems in RAL’s Ultracr facility – LIFEtime being one station – are exploited in studies ranging from fundamental reaction chemistry between hydrogen and a cyanide radical, to semiconductors and catalysts.
Complementary spectroscopy
Approaches that use probe pulses spanning from the UV to the mid-IR region include time-resolved fluorescence, transient electronic absorption, time-resolved IR and 2D IR, and Raman scattering. The DNA study is focusing on IR probe techniques because they provide the most detail about organic molecules. There is also a ‘whole bunch’ of other pump–probe techniques providing ‘information about how energy is shuffling between electronic and vibrational states within the molecules’, adds Towrie. ‘All of these techniques are complementary. No one of them gives you all the answers.’
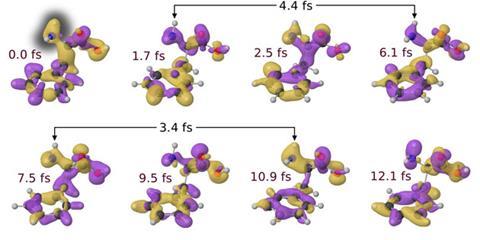
Helen Fielding from University College London in the UK is exploring the dynamics of electronic states in molecules excited by a UV pump laser pulse. Her team uses a second UV pulse as their probe, performing femtosecond time-resolved photoelectron spectroscopy, where they collect data from electron emissions triggered by the probe pulse. ‘The kinetic energy of the photoelectrons that are emitted provides us with information about the electronic and nuclear dynamics of the excited state,’ Fielding explains. ‘Their photoelectrons’ angular distribution can provide us with additional information about the electronic state that we are ionising.’
In this way the UCL group has explored the photochemistry of aniline, a structure found in many biomolecules, in particular how its excited state relaxes.2 Fielding and her colleague Jan Verlet from Durham University, UK, have also studied the chromophore from green fluorescent protein (GFP), a widely used biological label, in impressive detail. They found that after excitation the chromophore vibrates rapidly for around 100fs. Over the next 330fs a bond rotates, and then the molecule takes another 1.4ps to return to the ground state.3
Fielding echoes Towrie’s assertion that different techniques are complementary, and emphasises the relevance of the data they produce. ‘Together, they provide a more complete picture of the photoresponse of the molecules they are used to study,’ she explains. ‘The immediate beneficiaries are other scientists working in similar fields. However, understanding the photoresponse of molecules and complex systems at the microscopic scales is important in a diverse range of processes in nature and technology from light harvesting and therapies to nanoscale machines and electronic devices.’
Rulers of infinitesimal time and space
Vrakking, meanwhile, is striving to see processes that happen even more quickly. ‘When my pump laser starts a reaction, atoms start to change their position – but that isn’t the first thing that’s initiated,’ he explains. ‘There’s a very rapid coupling of electronic degrees of freedom and nuclear degrees of freedom.’ To explore what this causes, he is pushing the duration of his pump pulses down to the mind-bogglingly short attosecond (as) level.
An attosecond is a thousandth of a femtosecond, or a quintillionth of a second (10–18s). Yet its fleetingness is best evoked by the observation that there are roughly as many attoseconds in a second as there are seconds in the age of the universe. ‘The exciting thing about attosecond science is that we can go beyond atomic motion,’ Vrakking says. ‘The attosecond timescale give us access to electronic motion – that’s something that’s never been possible with femtosecond techniques.’
The ultimate dream is to be able to study systems down to the attosecond domain, at the same time with very good structural resolution
Working at this timescale involves accepting and working with the limits imposed by the Fourier transformation relationship. ‘For different types of experiments, we make this compromise differently,’ Vrakking admits. ‘Where I know I’m going to have sub-femtosecond electrodynamics, it will be absolutely essential to make an attosecond pulse.’
Necessarily starting with the simplest possible system – hydrogen – Vrakking’s team published the first attosecond pump–probe experiment on a molecule in 2010.4 Since then, scientists have been able to study more complex, biologically relevant, molecules. For example, in 2014 Mauro Nisoli at the Polytechnic University of Milan in Italy and colleagues used sub-300as pumps on the amino acid phenylalanine.5 The researchers then took snapshots using 4fs duration visible–NIR probes at delays of 500as and longer. And when they assembled the resulting ‘animation’, they could see charge transfer from one part of the molecule to another happened within that first 4.5fs.
Such insights clearly show the power that pump–probe techniques offer chemists – but these methods are poised to become still more remarkable. Attosecond pulses have to be in the extreme ultraviolet or even soft x-ray part of the electromagnetic spectrum, which brings additional opportunities, Vrakking underlines.
‘That means we can eject very energetic photoelectrons that in principle are capable of recognising their environment, a process where diffraction information can be encoded, as you are able to do with x-ray techniques,’ Vrakking says. ‘From such experiments people have already extracted bond distances, so attempts are definitely underway to try to combine the time resolution we have in attosecond experiments with structural information on the Ångstrom length scale. The ultimate dream is to be able to study systems down to the attosecond domain, at the same time with very good structural resolution.’
Thanks to pump–probe techniques, chemists are already routinely exploring a world whose pace and scale are completely alien – and they’re clearly not done yet. Dreams like Vrakking’s will drive the technology further into unexplored territory, helping researchers find scientific treasures as yet unknown.
Andy Extance is a science writer based in Exeter, UK
Article amended 05 May 2016 to correct John Kelly’s affiliation
References
1 J P Hall et al, Nat. Chem., 2015, 7, 961 (DOI: 10.1038/nchem.2369)
2 O M Kirkby et al, Phys. Chem. Chem. Phys., 2015, 17, 16270 (DOI: 10.1039/c5cp01883h)
3 R S Minns et al, Mol. Phys., 2013, 112, 1808 (DOI: 10.1080/00268976.2013.865809)
4 G Sansone et al, Nature, 2010, 465, 7299 (DOI: 10.1038/nature09084)
5 F Calegari et al, Science, 2014, 346, 336 (DOI: 10.1126/science.1254061)









No comments yet