Tamoxifen has been treating breast cancer for 40 years, but few would have predicted how much this drug would contribute to saving lives, writes John Mann
A woman’s lifetime risk of contracting breast cancer is one in eight, and we now know that oestrogens have a major influence on how this cancer develops. Although the sex hormones were not isolated and characterised until the 1930s, the young Scottish surgeon George Beatson believed that the development of breast cancer depended on a chemical stimulation from the ovaries. This presumption was supported when he removed the ovaries and fallopian tubes from a young patient with advanced breast cancer in June 1895, and seven months later reported1 that: ‘All vestiges of her previous cancerous disease had disappeared ` and she is apparently in excellent health.’
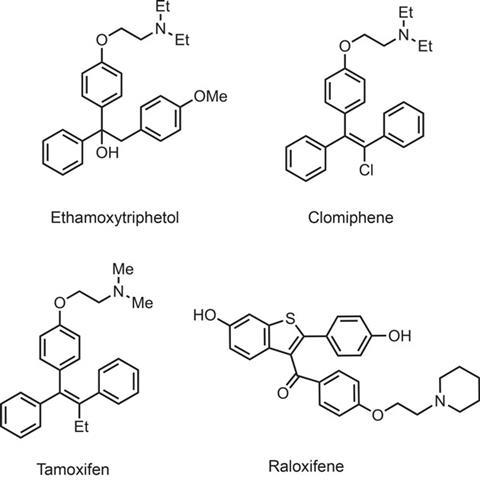
Drugs to treat breast cancer had to await the discovery of the steroid hormones, which was very much overshadowed by the excitement engendered by the contraceptive and anti-inflammatory steroids. In fact, the first true breast cancer drug, diethylstilbestrol, was originally developed as an oestrogen-like, or oestrogenic, agent, but was shown by Alexander Haddow in 1944 to help some breast cancer patients. Other analogues like ethamoxytriphetol and clomifene (developed by Merrell and ICI, respectively) were also designed as synthetic oestrogens but instead showed surprising anti-oestrogenic activities, with clomifene going on to become one of the key drugs used to induce ovulation and fertility in sub-fertile women. Hardly any interest was shown in the anti-cancer activities of these drugs since they exhibited unpleasant side effects when used under regular dosing regimes.
This all changed with the synthesis of tamoxifen by Dora Richardson at ICI’s Macclesfield, UK, site in 1962, as part of its fertility control programme. Michael Harper and Arthur Walpole, also of ICI, separated the two geometrical isomers and showed that the cis -form exhibited oestrogenic activity while the trans -form was an anti-oestrogen. Initially there was little enthusiasm within ICI for the drug since there were plenty of other fertility-regulating drugs on the market, but several small-scale clinical trials in 1971–73 clearly showed that tamoxifen produced good responses in women with advanced recurrent or metastatic breast cancer, and as a bonus there were no serious side-effects. However, in his review on 30 years of tamoxifen use, Craig Jordan (one of the pioneers of oestrogen receptor research) describes ‘a general clinical indifference to the development of a new endocrine agent for the treatment of advanced breast cancer’.
Nonetheless, Walpole managed to convince his superiors at ICI to take a chance on tamoxifen, and the drug was formally approved for wide-scale clinical use in 1973. Enthusiasm for its use increased when research revealed its mode of action through interaction with oestrogen receptors (ERs; see box below). It was thus a targeted drug, in contrast to the cytotoxic drugs that caused damage to both normal and cancer cells, and were at that time the drugs of choice for treating all cancers. Their modern counterparts, like doxorubicin, 5-fluorouracil and docetaxel, are still used where the breast cancer has spread to regional lymph nodes or beyond.
The huge impact of tamoxifen use was revealed by a report in The Lancet in January 1992, which reviewed a major study on 75,000 patients over a 10 year period.2 This showed that tamoxifen as a sole drug following surgical removal of a primary tumour led to a 25% reduction in recurrence of disease and a 17% reduction in mortality. In post-menopausal patients, tamoxifen and cytotoxic chemotherapy improved these figures to nearer 40%. What was even more remarkable was that prophylactic use in pre- and post-menopausal women at risk of developing breast cancer reduced incidence or recurrence dramatically. Although ICI patented tamoxifen as early as 1965 and identified that it ‘might be useful for the control of hormone-dependent tumours’, the US Food and Drug Administration (FDA) refused a US patent until 1985, after this prophylactic effect was first identified, effectively allowing ICI an extension of its exclusivity in the US for 17 years.
Oestrogen receptors
Elwood Jensen of the University of Chicago, US, discovered oestrogen receptors (ERs) on the surface of mammary cells in 1958, and he and others developed assays for these proteins. The hope that ER levels could predict the response to anti-oestrogens in breast cancer treatment was soon realised, and it is now known that ERs are overexpressed in more than 60% of mammary tumours.

There are two forms of ER and the a form is the most important in breast cancer. ERa dimerises after binding oestrogen or the 4-hydroxy metabolite of tamoxifen (the drug itself has a low affinity), and then translocates to the cell nucleus. In association with numerous protein cofactors, it binds to an oestrogen-responsive element on DNA. The complexity of these various interactions is still being clarified, but in essence oestrogen–ERa binding elicits recruitment of so-called p160 cofactors, while association between ERa and 4-hydroxytamoxifen recruits the co-repressor proteins NCOR1 and SMRT. Although oestrogen and 4-hydroxytamoxifen bind to the same a-helical pocket of the ERa, structural changes then reveal association sites for either the p160 cofactors or co-repressors respectively.
Once bound to the oestrogen response element on DNA, these complexes appear to control remodelling of the histone proteins that are associated with DNA (known as the chromatin complex). These lysine-rich proteins control the accessibility of DNA to transcription factors. Activation of histone acetyl transferases results in transfer of an acetyl group specifically to lysine groups with resultant neutralisation of the positive charges and consequent weakening of electrostatic DNA–histone interactions. This remodelling facilitates access of transcription factors to the DNA. Methylation mediated by histone methyltransferases effects similar remodelling. Tamoxifen disrupts these numerous interactions, and thus has a major therapeutic benefit for patients with oestrogen-dependent tumours.
Aromatase inhibitors
Once the importance of oestrogen had been identified, the search for agents to switch off oestrogen biosynthesis began. This has centred on inhibitors of the enzyme aromatase, which catalyses the conversion of the androgen hormones androstendione and testosterone into oestrone and oestradiol respectively. The enzyme is one of the cytochrome P450 class and is abundant in the placenta and ovaries, but is also present in the liver, subcutaneous fat, muscle, brain and both normal and cancerous breast cells. It is in these non-glandular tissues that enzyme activity resides after the menopause.
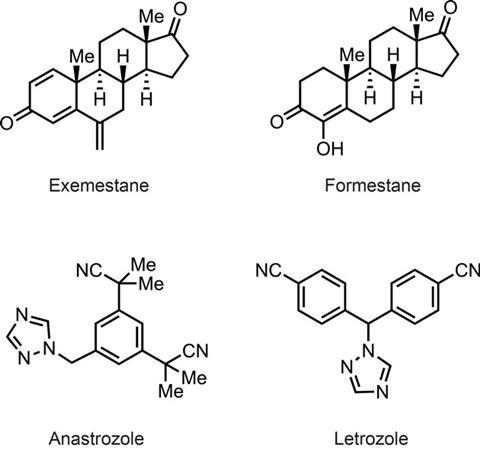
The drugs formestane and exemestane bind to the same site on the enzyme as androstendione, but are irreversible inhibitors of aromatase because they form reactive intermediates. Anastrozole (Arimidex) and letrozole (Femara) act as competitive inhibitors of aromatase by binding reversibly to the central haemin cofactor of the enzyme. These drugs are now favoured over tamoxifen for single agent use in post-menopausal patients, since they lead to total elimination of oestrogen production, rather than selective oestrogen receptor modulation (SERM). A recent paper in The Lancet3 reported that anastrozole has a prophylactic effect superior to that of tamoxifen when given to post-menopausal women at high risk of developing breast cancer. Furthermore, in a group of 4000 at-risk women in 18 countries, given either anastrozole or placebo, there were over 50% fewer breast cancers in the treatment group – a remarkable vote of confidence for this class of drugs.
HER2 overexpression
It has been known for years that some families have a greater susceptibility to breast cancer than others, suggesting a genetic involvement. The importance of gene over-expression or modification in breast cancer was first proved in the 1990s with the discovery that certain epidermal growth factor (EGF) receptors were over-represented on the cell surface in 20–30% of breast cancers. This was shown to result from overexpression of the HER2 gene. In normal cells, its activation promotes cell growth and division, but overexpression leads to uncontrolled cell growth and proliferation.
When an epidermal growth factor binds to the HER2 receptor on the outer face of the cell, this activates cell signalling pathways within the cell. This is mostly mediated by protein kinases such as MAPK (mitogen-activated protein kinase) and PI3K (phosphoinositol-3-kinase). These phosphorylate and activate other signalling molecules, induce various signalling cascades and ultimately induce cell division and angiogenesis (growth of new blood vessels). Consequently, a number of kinase inhibitors are now in clinical use, including lapatinib (Tyverb) and sunitinib (Sutent) to treat breast cancer.
But the major innovation in this area is use of the monoclonal antibody trastuzumab (Herceptin) which was developed in a collaboration between a large team at the University of California, Los Angeles, in the US and Genentech, and first approved by the FDA in September 1998. This binds to the HER2 receptor and leads to arrest of cell division and also suppresses angiogenesis. It extends survival by 20–25 months in late-stage metastatic breast cancer, and in early-stage breast cancer it reduces the absolute risk to around 10%.
There is also an apparent ‘cross-talk’ between the oestrogen receptor and HER2 signal pathways such that complexes of ERa and 4-hydroxytamoxifen repress HER2 gene transcription. This may occur because over-expression of HER2 is associated with increased phosphorylation of the ERa receptor and associated cofactors, and this apparently leads to enhanced access to transcription factors through chromatin remodelling.
Next in sequence
The other success story involves the genes BRCA1 and BRCA2, first cloned in 1994·5. These code for proteins that function as tumour-suppressors and are involved in repairing breakages in double-stranded DNA, but also appear to inhibit cell proliferation by stabilising DNA–histone complexes. 10–15% of heritable breast cancers have a mutation in BRCA1 or BRCA2. Patents covering the gene sequences for BRCA1 and BRCA2 were granted in 1997–8 to Myriad Genetics of Salt Lake City, US, in collaboration with the University of Utah and others, with the intention of controlling access to gene assays and to drugs that regulated gene expression.
The patents were contested in 2010 and, after much legal wrangling, they were finally invalidated by the US Supreme Court in June 2013, which ruled that human genes are ‘products of nature’ and hence cannot be patented. However, patents covering the various assays for the genes are still valid, and access to these assays is the key to therapy, since mutation in one or both genes predisposes to early onset breast (and ovarian) cancer. Women who have a positive analysis for these mutations may choose to have prophylactic surgery or use anti-oestrogens, and this should reduce the lifetime risk of breast cancer
Other genes that are mutated in more than 10% of breast cancers include TP53, PI3KCA, MAP3K1 and GATA3. The first of these are the genes that code for the production of the tumour suppressor proteins p53. These are often called the ‘guardians of cells’ since they co-ordinate DNA repair, or if this fails they shut off cell division and initiate the process called apoptosis (controlled cell death). PI3KCA and MAP3K1 are two of the key kinases involved in cell signalling; GATA3 codes for an important transcription factor. However, the sheer complexity of gene mutations in breast cancer (as many as 1700 different mutations identified thus far) suggests that the quest for gene specific therapy is still a long-term goal.
40 years on
Tamoxifen has undoubtedly improved the survival chances of millions of women during the past 40 years. However, the long-term use of tamoxifen is now thought to increase the risk of cancer of the womb lining in post-menopausal patients, and use of the newer anti-oestrogen raloxifene (Evista) and aromatase inhibitors is now favoured. A comparison of the prophylactic effects of raloxifene versus tamoxifen in high risk patients (around 19,750 patients over five years, reported in 2006) was identical, with about 50% less incidence of breast cancer, but there were nearly 40% fewer uterine cancers and 30% fewer blood clots with raloxifene. It also reduces osteoporosis in post-menopausal patients, and this unexpected benefit is due to the drug’s remarkable selective oestrogen activity in bone versus its anti-oestrogenic activity in the breast and uterus. Other SERM agents with even greater selectivity are undergoing clinical evaluation.
Tamoxifen has spawned numerous lines of investigation, and has moved clinicians closer to the goal of identifying targeted drug regimes that are appropriate for individual patients. Nonetheless, it is sobering to note that there are still 1.3 million new cases of breast cancer worldwide each year with 450,000 deaths, and the lifetime risk of a woman developing breast cancer is still one in eight. In the continuing quest for new and better drugs, complacency is certainly not an option.
John Mann is author of the book Life Saving Drugs, published by the RSC
References
1 G T Beatson, Lancet, 1896, 148, 104 (DOI: 10.1016/S0140-6736(01)72307-0)
2 Early breast cancer trialists’ collaborative group, Lancet, 1992, 339, 71 (DOI: 10.1016/0140-6736(92)90997-H)
3 J Cuzick et al, Lancet, 2013, DOI: 10.1016/S0140-6736(13)62292-8












No comments yet