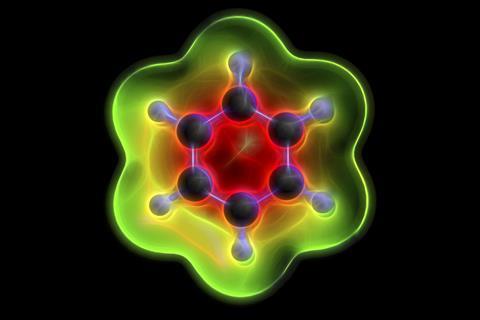
Scientists in South Korea are reporting the most accurate experimental geometries for benzene and deuterated benzene to date.1 The team led by Thomas Schultz at the Ulsan National Institute of Science and Technology say that previously reported experimental structures were out by several milliangstroms.
Sérgio Domingos from the University of Coimbra in Portugal, who wasn’t involved in the study, explains that isotopic information was used to determine the corrected benzene structure. ‘The results reveal carbon–hydrogen and carbon–deuterium bond lengths that defy currently accepted literature values,’ he says. ‘The nuclear quantum effect of deuterium with respect to hydrogen hasn’t been explored for benzene with this level of precision before.’
Benzene was first isolated by Michael Faraday almost 200 years ago and has become an important reference molecule that is used to test experiments and theoretical methods in different fields. But despite being studied intensively, benzene’s behaviour continues to surprise scientists. Recently, a high-resolution rotational spectroscopy study revealed an unusual hydrogen/deuterium isotope effect2 as well as unexpected differences between the effective and equilibrium carbon–hydrogen bond lengths of this compound. The new experimental work challenges these observations, suggesting that benzene’s vibrational properties are just like those of other small molecules.
Schultz and colleagues mention that their results ‘refute recent experimental and theoretical literature claims of identical effective C–H and C–D bond lengths in benzene’, and indeed the team found an 11.5mÅ difference between the two, with the deuterium bond shorter. ‘The researchers have identified shortcomings of earlier approximations which led to surprising results for this prototypical aromatic molecule,’ comments Jennifer van Wijngaarden from York University in Canada. ‘The agreement with quantum chemical theory is impressive and these results are more intuitive than what the prior microwave studies suggested.’
Domingos notes that the technique used – mass-correlated rotational alignment spectroscopy – is a tandem one that correlates the rotational Raman spectra of neutral ground-state molecules with the masses of ions formed during ionisation. ‘The method allows for complicated isotopic species to be studied, even molecules with a lot of symmetry which are often not detectable with other techniques,’ he says.
The approach uses a pulse from a pump laser to create excited rotational states of molecules using Raman excitation. ‘Rather than detecting the rotational frequency directly to identify these excited states, the detection is done by ionising the rotating molecules using two different photons to reach sufficient energy, one fixed and one variable,’ explains van Wijngaarden. She says that the new study corrects current understanding of the structure of benzene and lays the framework for building better models of molecular structure. ‘And as this technique can be applied to highly symmetric molecules, it opens the door to the characterisation of many chemically important species like polycyclic aromatic hydrocarbons.’
References
1 I Heo et al, J. Phys. Chem. Lett., 2022, 13, 8278 (DOI: 10.1021/acs.jpclett.2c02035)
2 S Kunishige et al, J. Chem. Phys., 2015, 143, 244302 (DOI: 10.1063/1.4937949)

![An image showing a [M2(BzN6-Mes)]n− complex](https://d2cbg94ubxgsnp.cloudfront.net/Pictures/380x253/4/0/8/514408_index_128835.jpg)








4 readers' comments