Stubbornly stable unactivated internal alkenes become chiral tertiary amine precursors
Some carbon-carbon double bonds seem too unreactive for synthetic use – but that’s just how chemists in the US are now exploiting them. Stephen Buchwald’s Massachusetts Institute of Technology (MIT) team’s copper-catalysed process adds secondary amine groups and hydrogen atoms across double bonds of unactivated internal alkenes.1
‘We were surprised that compounds we normally think unreactive work so well,’ Buchwald tells Chemistry World. Although 20,000 tonnes of the simplest unactivated internal alkene, 2-butene, are produced annually the MIT researcher doesn’t see the new reaction turning it into commodities yet. ‘I think it’ll be used for doing pharmaceutical discovery,’ he predicts.
The process builds on growing knowledge of making amines from molecules like alkenes using copper (I) hydride complexes. In the MIT chemists’ catalytic cycle, copper hydride first inserts into the alkene’s double bond, with the copper atom and its ligands remaining covalently attached to the product. This molecule couples with a hydroxylamine ester, whose secondary amine displaces the copper, which in turn bonds to the ester group. A hydrogen atom from a silane reagent then displaces the ester, regenerating the copper hydride.
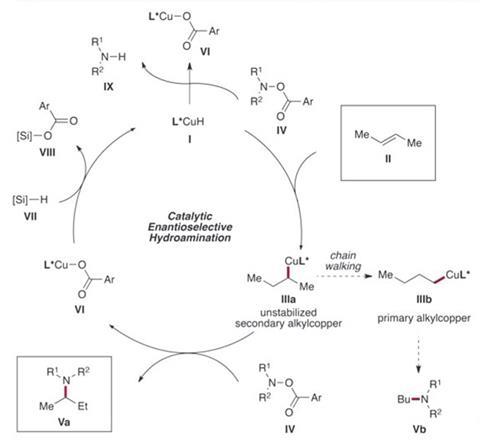
However, the copper hydride can reduce the hydroxylamine ester instead, especially if the alkene reduction is slow. Success therefore relies in part on complexing the copper hydride with the right ligand partner. In the Buchwald team’s first copper hydride chemistry work, published in 2013, Shaolin Zhu screened five ligands’ effectiveness in making amines from the comparatively reactive alkene styrene.2 The highest yields and enantioselectivity came with one known as DTBM-SEGPHOS, already known to impart a strong chirality preference to reactions using copper (I) hydride. ‘That gave us the best results, and we’re trying to understand why,’ Buchwald says.
Great challenge
Buchwald’s group had previously identified using unactivated internal alkenes in synthesis as one of the ‘greatest challenges’ they could pursue. Team-members Yang Yang and Shiliang Shi therefore tried it with the DTBM-SEGPHOS copper complex, producing an amine in 34% yield and 98% enantiomeric excess (ee), much to Buchwald’s excitement. ‘These people will, within reason, try anything no matter how daunting, so I give them all the credit,’ he says.
The chemists subsequently optimised the reaction, for example varying the hydroxylamine ester to minimise any unwanted reduction and speed up the reaction, and then explored how general it was. They made a wide variety of chiral amines with trans-but-2-ene in yields over 50% while retaining high ees, and showed many unactivated internal olefins could also be used.
John Brown, who researches catalysis for enantioselective organic synthesis at the University of Oxford, UK, emphasises how the reaction’s selectivity builds on the team’s previous work. ‘Their catalyst and conditions avoid competing isomerization of the double bond so that the position of the new stereocentre is defined,’ he explains.
‘Methods that make important chiral compounds selectively from cheap and ubiquitous building blocks like alkenes have particularly high potential,’ says Matt Clarke from the University of St Andrews. Clarke, who also studies asymmetric synthesis and catalysis, calls the new approach ‘a very nice example’ of such a method. ‘If you wanted to make a chiral tertiary amine in the lab, then this must be right up there with any other methods. At seriously large scale, reagents and ligand would probably cost too much in its present form, but clearly this is a promising approach.’












No comments yet