The molecule 5-methyl-2-[(2-nitrophenyl)amino]-3-thiophenecarbonitrile, commonly called Roy (red–orange–yellow) because of the colours of its crystals, is known as the organic compound with the most polymorphs. Now, Gregory Beran and collaborators from the University of California Riverside, US, and Imperial College London, UK, have developed the first crystal energy landscape for Roy that agrees with experimental evidence and concluded that researchers have already found the most stable Roy polymorphs.1
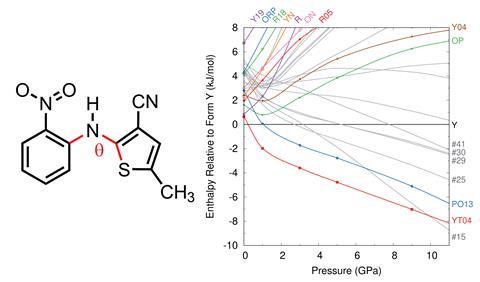
Previously, crystal structure predictions have not shown the same relative stability that is seen through experimentation. Density functional theory (DFT) is known to overcorrect for stability due to delocalisation errors in systems with greater π-conjugation, such as planar molecules. A conformational energy-correction can be applied to DFT to reduce this error, which, when combined with previous crystal structure predictions, results in a more accurate energy landscape reflecting the experimental evidence.
The new landscape developed by Beran and colleagues shows the seven most stable polymorphs of Roy under ambient conditions have already been discovered. Whilst there is potential for new polymorphs to be reported under these conditions, the landscape suggests specialised crystallisation techniques may be required. However, at pressures in excess of 10GPa, it identified an as yet unreported polymorph to be more stable than the rest.
Walter McCrone famously stated that, ‘every compound has different polymorphic forms, and that, in general, the number of forms known for a given compound is proportional to the time and money spent in research on that compound.’2 This new study shows how the limits of conventional computational chemistry can be overcome to give more accurate crystal structure predictions.
References
1 G Beran et al, Chem. Sci., 2022, DOI: 10.1039/d1sc06074k (This article is open access.)
2 W C McCrone, Polymorphism, Physics and Chemistry of the Organic Solid State, Vol 2, pp. 725–767, New York, Wiley Interscience, 1965










1 Reader's comment